Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
(Mark One)
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the Fiscal Year Ended December 31, 2008
or
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Commission File Number: 000-29959
Pain Therapeutics, Inc.
(Exact name of registrant as specified in its charter)
| Delaware | 91-1911336 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification Number) |
2211 Bridgepointe Parkway
Suite 500
San Mateo, CA 94404
(650) 624-8200
(Address, including zip code, or registrant’s principal executive offices and
telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act: Common Stock, $0.001 par value
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definition of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer ¨ | Accelerated filer x | |
| Non-accelerated filer ¨ | Smaller reporting company ¨ | |
| (Do not check if a smaller reporting company) |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2). Yes ¨ No x
The aggregate market value of the voting and non-voting common equity held by non-affiliates was $419,667,047 computed by reference to the last sales price of $7.90 as reported on the NASDAQ Global Market, as of the last business day of the Registrant’s most recently completed second fiscal quarter, June 30, 2008.
The number of shares outstanding of the Registrant’s common stock on January 12, 2009 was 42,078,231.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the Registrant’s Proxy Statement for its 2009 Annual Meeting of Stockholders (the “Proxy Statement”), to be filed with the Securities and Exchange Commission, are incorporated by reference to Part III of this Form 10-K Report.
Table of Contents
FORM 10-K
INDEX
| Page | ||||
| PART I | ||||
| Item 1. |
2 | |||
| Item 1A. |
10 | |||
| Item 1B. |
25 | |||
| Item 2. |
25 | |||
| Item 3. |
25 | |||
| Item 4. |
25 | |||
| PART II | ||||
| Item 5. |
Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities | 26 | ||
| Item 6. |
28 | |||
| Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
29 | ||
| Item 7A. |
37 | |||
| Item 8. |
38 | |||
| Item 9. |
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
58 | ||
| Item 9A. |
58 | |||
| Item 9B. |
60 | |||
| PART III | ||||
| Item 10. |
60 | |||
| Item 11. |
60 | |||
| Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | 60 | ||
| Item 13. |
Certain Relationships and Related Transactions and Director Independence |
61 | ||
| Item 14. |
61 | |||
| PART IV | ||||
| Item 15. |
61 | |||
Table of Contents
This document contains forward-looking statements that are based upon current expectations that are within the meaning of the Private Securities Reform Act of 1995. We intend that such statements be protected by the safe harbor created thereby. Forward-looking statements involve risks and uncertainties and our actual results and the timing of events may differ significantly from the results discussed in the forward-looking statements. Examples of such forward-looking statements include, but are not limited to statements about:
| • |
submission of additional non-clinical data to the U.S. Food and Drug Administration, or FDA, to support the potential approval of the New Drug Application, or NDA, for Remoxy®; |
| • | royalty, milestone or collaboration revenue we may receive from King Pharmaceuticals, Inc., or King, and other payments we may receive from our strategic alliances; |
| • | the duration of the development period for all four expected drug candidates under our collaboration with King; |
| • | anticipated clinical trials and preclinical studies; |
| • | potential sources of commercial supply of Remoxy and its components; |
| • | expansion of our potential product line, including the formulation of additional dosage forms of our drug candidates; |
| • | future operating losses and anticipated operating and capital expenditures; |
| • | uses of proceeds from our securities offerings; |
| • | the potential benefits of our drug candidates; |
| • | the sufficiency of materials required for the clinical development of our drug candidates; |
| • | the size of the potential market for our products; |
| • | the utility of protection of our intellectual property; |
| • | expected future sources of revenue and capital and increasing cash needs; |
| • | potential competitors or competitive products; |
| • | future market acceptance of our drug candidates and potential drug candidates; |
| • | expenses increasing or fluctuations in our operating results; |
| • | future expectations regarding trade secrets, technological innovations, licensing agreements and outsourcing of certain business functions; |
| • | anticipated hiring and development of our internal systems and infrastructure; |
| • | the sufficiency of our current resources to fund our operations over the next twelve months; |
| • | assumptions and estimates used for our disclosures regarding stock-based compensation; and |
| • | estimates concerning the provision for (benefit from) income taxes and realization of deferred tax assets. |
Such forward-looking statements involve risks and uncertainties, including, but not limited to, those risks and uncertainties relating to:
| • | the possibility of the FDA requesting additional non-clinical data which would significantly delay or prevent the potential approval of Remoxy; |
| • | the successful development and commercialization of Remoxy and other drug candidates pursuant to our collaboration agreement with King and development of other drug candidates pursuant to our other collaboration agreements, and the continuation of such agreements; |
| • | difficulties or delays in development, testing, clinical trials (including patient enrollment), regulatory approval, production and commercialization of our drug candidates; |
1
Table of Contents
| • | unexpected adverse side effects or inadequate therapeutic efficacy of our drug candidates that could slow or prevent product approval (including the risk that current and past results of clinical trials are not indicative of future results of clinical trials) or potential post-approval market acceptance; |
| • | the uncertainty of protection of our intellectual property rights or trade secrets; |
| • | potential infringement of the intellectual property rights of third parties; |
| • | pursuing in-license and acquisition opportunities; |
| • | maintenance or third party funding of our collaboration and license agreements; |
| • | hiring and retaining personnel; and |
| • | our financial position and our ability to obtain additional financing if necessary. |
In addition, such statements are subject to the risks and uncertainties discussed in the “Risk Factors” section and elsewhere in this document.
| Item 1. | Business |
Overview
We are a biopharmaceutical company that develops novel drugs. In June 2008, we submitted an NDA for Remoxy with the FDA. In August 2008, we and King announced that an NDA for Remoxy was accepted by the FDA and granted Priority Review, which generally results in a reduction in the time for FDA review of an NDA from 12 months to approximately 6 months from the date of the NDA submission. In December 2008, we received a Complete Response Letter for our NDA for Remoxy in which the FDA determined that the NDA was not approved. The FDA indicated additional non-clinical data will be required to support the approval of REMOXY. The FDA has not requested or recommended additional clinical efficacy studies prior to approval. We plan to meet with the FDA in the second quarter of 2009 regarding the NDA for Remoxy. We believe this FDA meeting will provide us with a more reliable context in which to make projections about Remoxy.
We also have the following investigational drug candidates in clinical programs:
| • | PTI-202 and PTI-721, which are proprietary, abuse-resistant forms of opioid drugs. |
| • | A novel radio-labeled monoclonal antibody drug candidate to treat metastatic melanoma, a deadly form of skin cancer. |
We and King are engaged in a strategic alliance to develop and commercialize Remoxy, PTI-202, PTI-721 and another abuse-resistant opioid painkiller. King is also obligated to fund certain development expenses incurred by us pursuant to the collaboration agreement.
Remoxy
Remoxy is a novel controlled-release oral capsule form of oxycodone in a highly viscous liquid formulation matrix that includes novel excipients. It is specifically formulated to help address issues of abuse and misuse of time-release oxycodone tablets. Sales of time-release oxycodone were estimated to be over $2.0 billion in 2008.
The analgesic action of extracts of the opium poppy plant has been known for millennia. In more recent decades, semi-synthetic opium derivatives, such as oxycodone, a generic drug in clinical use since the 1930’s, have become a standard of care for treating moderate-to-severe pain.
It is also well-known that medicinal opioids such as oxycodone can produce both analgesia and euphoria. The search for euphoric effects can lead to drug-seeking behavior, tolerance and dependence. In particular, rapid increases in plasma levels of oxycodone may lead to overdose, respiratory depression or death.
2
Table of Contents
Opioid misuse and abuse are significant public health problems. The active drug ingredient in time-release oxycodone is oxycodone, an FDA approved substance for the relief of moderate to severe pain. Oxycodone is generally considered safe and effective when properly prescribed, dispensed and administered for legitimate medical purposes.
However, the U.S Drug Enforcement Administration, or DEA, has reported that time-release oxycodone tablet abuse has substantially increased since FDA approval of time-release oxycodone tablet abuse. Abusers can quickly and easily extract large amounts of oxycodone by simply breaking or crushing time-release oxycodone tablets. Doing so disrupts this time release mechanism and allows an abuser to immediately ingest, snort or inject a large dose of oxycodone that was originally intended to be slowly release over 12 hours. Rapid increases in plasma levels of oxycodone may lead to overdose, respiratory depression or death. Patients may mistakenly cut or crush the time-release oxycodone tablets, which may also lead to accidental overdose.
The Remoxy formulation is designed to resist common methods of chemical or physical manipulation. Remoxy’s capsule dosage form provides therapeutic drug levels of oxycodone on a twice-daily dosing schedule, while resisting the rapid increases in plasma levels of oxycodone associated with common methods of abuse and misuse. Its formulation also resists delivery by unapproved routes of administration, such as injection, snorting or inhalation.
Remoxy is intended to meet the needs of physicians who appropriately prescribe opioid painkillers and who seek to minimize the risks of drug diversion, abuse or accidental patient misuse as well as the needs of pharmacists and the managed care healthcare system in the United States.
Other abuse resistant product candidates
We believe the abuse-resistant technology used in Remoxy is applicable to different oral opioid painkillers. Our strategic alliance with King includes three other product candidates. PTI-202 and PTI-721 have completed Phase I clinical trials. The active ingredients in PTI-202 and PTI-721 are opioids whose identities have not been announced. These Phase I clinical trials were designed to investigate the safety, tolerability, pharmacokinetics and pharmacodynamic profile of a single, oral dose of the drug candidates in healthy volunteers. We believe results also indicate these product candidates are safe and well-tolerated and their release profile appears well-suited to use with a chronic pain population. Like Remoxy, PTI-202 and PTI-721 are intended to meet the needs of physicians who appropriately prescribe opioid painkillers and who seek to minimize the risks of drug diversion, abuse or accidental patient misuse. King made milestone payments to us of $5.0 million in 2008 in connection with the acceptance by the FDA of the investigational new drug application, or IND, for PTI-721 and $5.0 million in 2006 in connection with the acceptance by the FDA of the IND for PTI-202. We and King are also conducting pre-clinical development of a fourth product candidate.
Metastatic Melanoma
Our monoclonal antibody program, developed at Albert Einstein College of Medicine, is aimed at treating patients with metastatic melanoma. These monoclonal antibodies deliver lethal radiation to melanoma tumors. The technology may also have therapeutic uses outside of oncology, such as infectious diseases. We licensed exclusive worldwide commercial rights to this technology from Albert Einstein College of Medicine.
We believe that this antibody technology may enable clinicians to provide effective medical treatment for metastatic melanoma, a deadly form of skin cancer and that the specificity of this treatment may be effective against tumors without harming normal tissue.
In June 2008, we announced the successful completion of our first clinical study using the radio-labeled monoclonal antibody technology. The objectives of this study were to assess safety, pharmacokinetics and
3
Table of Contents
dosimetry. Top-line results of this clinical study indicate this antibody binds to melanoma tumor sites. No drug- related serious adverse events were observed in this study. In 2009, we plan to initiate a second clinical study in which patients will receive increasing amounts of the radio-labeled antibody. We expect to complete the second clinical study in the second half of 2009.
The antibody technology being developed for metastatic melanoma may also have therapeutic uses outside of oncology. We are conducting pre-clinical studies using this technology in certain infectious diseases. We plan to continue these pre-clinical studies in 2009.
Hemophilia
Our gene transfer program, developed at Stanford University, is aimed at correcting an underlying genetic defect in patients with hemophilia. Sometimes known as ‘the bleeding disease’, hemophilia affects 400,000 people worldwide with average treatments ranging between $60,000 and $150,000 per patient/year. We licensed this technology from Poetic Genetics, LLC, or Poetic. During 2008, we conducted a variety of pre-clinical studies with this technology. We expect to complete an animal study with this technology in 2009.
Strategy
Our goal is to continue to develop novel drugs that are more effective or safer than drugs used in the clinic today. Our strategy includes the following elements:
Focus on Clinical Development Stage Products. We believe this focus will enable us to generate product revenues earlier than if we were focused on early-stage research and discovery activities.
Retain Significant Rights to Our Drugs. We currently retain worldwide commercialization rights to all of our technology and drug candidates in all markets and indications, except for Remoxy and certain other abuse-resistant drugs that are subject to our strategic alliance with King. In general, we intend to independently develop our drug candidates through late-stage clinical trials. In market segments that require large or specialized sales forces, we may seek sales and marketing alliances with third parties.
Outsource Key Functions. We intend to continue to outsource preclinical studies, clinical trials and formulation and manufacturing activities. We believe outsourcing permits significant time savings and allows for more efficient deployment of our resources.
Pursue In-licensing or Acquisition Opportunities. We intend to evaluate promising drug candidates or technologies to further expand our product pipeline. Our in-licensing strategy consists of evaluating clinical or preclinical stage opportunities in therapeutic areas that can benefit from our core expertise in drug development. Such in-licensing or acquisition opportunities may be in pain management or in other therapeutic areas outside of pain management. We believe this element of our corporate strategy could diversify some of the risks inherent in focusing on a single therapeutic area and could also increase our probability of commercial success.
We also conduct basic research in collaboration with academic and other partners. Company sponsored research and development expenditures were $46.8 million, $47.7 million and $45.8 million in 2006, 2007, and 2008 respectively. We recorded contract revenue of $22.7 million, $42.7 million $29.4 million in 2006, 2007, and 2008 respectively, related to customer-sponsored research activities under our collaboration with King.
Our Intellectual Property
We seek to protect our technology by, among other methods, filing and prosecuting U.S. and foreign patents and patent applications with respect to our technology and products and their uses. The focus of our patent strategy is to secure and maintain intellectual property rights to technology for the following categories of our business:
| • | the technology that forms the basis of Remoxy and our other abuse resistant drug candidates drug candidates; |
4
Table of Contents
| • | the clinical use of an ultra-low-dose opioid antagonist, either alone or in combination with an opioid painkiller, for pain management and opioid and other addiction; |
| • | the use of an ultra-low-dose opioid antagonist to render opioid-based products more effective; |
| • | the clinical use of an ultra-low-dose opioid antagonist, either alone or in combination with any opioid painkiller, for the treatment of other conditions; |
| • | the clinical use of radio-labeled monoclonal antibodies for the treatment of metastatic melanoma and certain therapeutic uses outside of oncology; |
| • | the clinical uses of a unique gene integration system intended to treat hemophilia or pain; |
| • | the technologies or intellectual property related to our pre-clinical product candidates; and |
| • | the manufacture and use of our drug candidates. |
We plan to prosecute and defend our patent applications, issued patents and proprietary information. Our competitive position and potential future revenues will depend in large part upon our ability to protect our intellectual property from challenges and to enforce our patent rights against potential infringements.
We and our collaborators have filed patent applications with the U.S. Patent Office to further protect our technologies. Certain patents are issued and a number of patent applications are pending. If issued, we believe these applications would protect certain of our technologies through at least 2020. If these patent applications do not result in issued patents, the duration or scope of our patent rights may be limited and our future revenues could be lower as a result.
If our competitors are able to successfully challenge the validity or scope of our patent rights, based on the existence of prior art or otherwise, they might be able to market products that contain features and clinical benefits similar to those of our drug candidates, and demand for our drug candidates could decline as a result.
We may be involved in additional challenges to our intellectual property. An adverse outcome of any challenges to our intellectual property could result in loss of claims of these patents that pertain to certain drugs we currently have under development and could have a material adverse impact on our future revenues.
Strategic Alliance with King Pharmaceuticals, Inc.
We have a collaboration agreement and a license agreement with King to develop and commercialize Remoxy and other abuse-resistant opioid painkillers. King made an upfront cash payment of $150.0 million to us at the closing of this strategic alliance in 2005.
In 2008, we received $20.0 million in cumulative milestone payments from King for achieving clinical and regulatory milestones, including $15.0 million related to acceptance by the FDA of the NDA for Remoxy, and $5.0 million of acceptance by the FDA of the IND for PTI-721. In 2006, we received $5.0 million for the acceptance by the FDA of the IND for PTI-202. We could also receive from King up to $125.0 million in additional milestone payments in the course of clinical development of Remoxy and other abuse-resistant opioid painkillers under the strategic alliance. In addition, subject to certain limitations, King is obligated to fund development expenses incurred by us pursuant to the collaboration agreement.
Pursuant to the license agreement, King is obligated to fund the commercialization expenses of, and has the exclusive right to market and sell, drugs developed pursuant to the strategic alliance, and is obligated to pay us a 20% royalty on net sales, except as to the first $1.0 billion in cumulative net sales, for which the royalty is set at 15%. King is also obligated to reimburse us for our payment of third-party royalty obligations related to this strategic alliance.
5
Table of Contents
We formed a joint oversight committee, or JOC, with King to oversee drug development and commercialization strategies for the strategic alliance. Pursuant to the collaboration agreement in the strategic alliance, we retain sole control of drug development activities in the United States through Phase II clinical trials. We and King will jointly manage Phase III clinical trials and NDA submissions in the United States. King has responsibility for these development activities outside the United States. Upon regulatory approval, King will assume sole control and worldwide responsibility to exclusively commercialize abuse-resistant opioid drugs developed pursuant to the strategic alliance. We retain all development and commercial rights in Australia and New Zealand.
The collaboration agreement continues until the later of the expiration of any patent rights licensed under the license agreement or developed under the collaboration agreement and the expiration of all periods of market exclusivity with respect to Remoxy and other abuse-resistant opioid drug candidates being developed under the strategic alliance. We and King can terminate the collaboration agreement under certain circumstances, including material breach and insolvency. King can terminate the collaboration agreement six months after the third anniversary of the effective date of the collaboration agreement, or sooner if the JOC determines that the development program under the collaboration agreement is unlikely to generate any marketable products. Our license agreement with King terminates at the time that the collaboration agreement terminates.
Formulation Agreement with Durect Corporation
We have an exclusive, worldwide licensing agreement with Durect Corporation, or Durect, to use a patented technology that forms the basis for a number of oral gel-cap drug candidates, including Remoxy. We have sub-licensed to King certain rights to develop and to commercialize Remoxy and certain other opioid drugs formulated in part with technology we licensed from Durect. Under the agreement with Durect, we control all of the preclinical, clinical, commercial manufacturing and sales/marketing activities for Remoxy and other abuse-resistant opioid painkillers. We reimburse Durect for formulation and related work, and make milestone payments based on the achievement of certain technical, clinical or regulatory milestones. Durect will supply us with certain components of Remoxy and other abuse-resistant opioid painkillers on a cost-plus basis. We also are obligated to pay Durect royalties on any related drug sales. In turn, King is obligated to reimburse us for costs we incur under the agreement with Durect, including milestone payments and royalties.
Under our license agreement with King, we are obligated not to amend or terminate our agreement with Durect if an amendment or termination would alter the rights or obligations of King under the collaboration agreement or license agreement.
License of Technology from Albert Einstein College of Medicine
We have licensed certain technology, including antibody technology, from Albert Einstein College of Medicine. We have a worldwide exclusive license to the technology and all intellectual property rights arising from the technology. Pursuant to the agreements for the licensed technology, we are required to pay Albert Einstein College of Medicine clinical milestone payments and royalties based on a percentage of net drug sales. If a product is combined with a drug or other substance for which we are paying an additional royalty, the royalty that we pay to Albert Einstein College of Medicine will be reduced by up to one-half of the amount of such additional royalty.
Albert Einstein College of Medicine originally received grants from the U.S. federal government to research some of the technology that we license. The terms of these grants provide the U.S. federal government with a non-exclusive, non-transferable paid-up license to practice inventions made with federal funds. Thus, our licenses are non-exclusive to the extent of the U.S. federal government’s license. If the U.S. federal government exercises its rights under this license, it could make use of the same technology that we license and the size of our potential market could thereby be reduced.
6
Table of Contents
License of Technology from Poetic Genetics, LLC
We have licensed novel gene integration technology from Poetic. We have worldwide commercial rights and all intellectual property rights arising from the technology in all indications in hemophilia and pain management. In connection with the license, we paid Poetic an undisclosed upfront fee and we are obliged to pay Poetic milestone payments of up to $4 million in the aggregate, based on clinical and regulatory progress, and a 4% royalty on net sales, or 6% if the first U.S. sale of a licensed product to treat hemophilia occurs before January 1, 2011.
Manufacturing
We do not own any manufacturing facilities. We plan to continue to outsource formulation, manufacturing and related activities.
We rely on a limited number of third-party manufacturers to formulate, manufacture, fill, label, ship or store all of our drug candidates. We have entered into agreements with and rely upon qualified third parties for the formulation or manufacture of our clinical supplies. These supplies and the manufacturing facilities must comply with DEA regulations and current good manufacturing practices, or GMPs, enforced by the FDA and other government agencies.
We and King rely on Durect and other third-party manufacturers to formulate, manufacture, fill, label, ship or store Remoxy and other abuse-resistant drug candidates and their components. King is responsible for commercial manufacturing and supply of Remoxy. Remoxy and other product candidates under our strategic alliance with King are formulated using, in part, proprietary technology licensed from Durect. Remoxy uses bulk oxycodone supplied by Noramco, Inc.
Government Regulation
Regulation by governmental authorities in the United States and other countries is a significant factor in the manufacture and marketing of pharmaceuticals and in our ongoing research and development activities. All of our products will require regulatory approval by governmental agencies prior to commercialization. In particular, human therapeutic products are subject to rigorous preclinical testing and clinical trials and other pre-marketing approval requirements by the FDA and regulatory authorities in other countries. In the United States, various federal, and in some cases state, statutes and regulations also govern or impact upon the manufacturing, safety, labeling, storage, record keeping and marketing of our products. The lengthy process of seeking required approvals and the continuing need for compliance with applicable statutes and regulations require us to spend substantial resources. Regulatory approval, when and if obtained, may be limited in scope which may significantly limit the indicated uses for which our products may be marketed. Further, approved drugs, as well as their manufacturers, are subject to ongoing review and discovery of previously unknown problems with such products that may result in restrictions on their manufacture, sale or use or in their withdrawal from the market. For example, on February 9, 2009, the FDA announced it had invited the sponsors of opioid drug products to a private meeting in March 2009 in order to begin the process of developing Risk Evaluation and Mitigation Strategies for opioid drugs. According to FDA’s announcement, opioid drugs affected by this announcement include both brand name and generic products that contain fentanyl, hydromorphone, methadone, morphine, oxycodone and oxymorphone.
Applicable FDA regulations require the filing of an NDA or a Biologic License Application, or BLA and approval by the FDA prior to commercialization of any of our drug candidates in the United States.
The Drug Approval Process
We will be required to complete several activities before we can market any of our drug candidates for human use in the United States, including:
| • | preclinical studies; |
7
Table of Contents
| • | submission to the FDA of an IND which must become effective before human clinical trials commence; |
| • | adequate and well-controlled human clinical trials to establish the safety and efficacy of the drug candidate; |
| • | submission to the FDA of an NDA; and |
| • | FDA approval of the NDA prior to any commercial sale or shipment of the drug. |
Preclinical tests include laboratory evaluation of product chemistry and formulation, as well as animal studies to assess the potential safety of the product. Preclinical safety tests must be conducted by laboratories that comply with FDA regulations regarding Good Laboratory Practice. We submitted the results of preclinical tests to the FDA as part of our INDs prior to commencing clinical trials. We may be required to conduct additional toxicology studies.
Based on preclinical testing, an IND is filed with the FDA to begin human testing of the drug in the United States. The IND becomes effective if not rejected by the FDA within 30 days. The IND must indicate the results of previous experiments, how, where and by whom the new clinical trials will be conducted, the chemical structure of the compound, the method by which it is believed to work in the human body, any toxic effects of the compound found in the animal studies and how the compound is manufactured. All clinical trials must be conducted in accordance with Good Clinical Practice. In addition, an Institutional Review Board, or IRB, generally comprised of physicians at the hospital or clinic where the proposed clinical trials will be conducted, must review and approve the IND. The IRB also continues to monitor the clinical trial. We must submit progress reports detailing the results of the clinical trials to the FDA at least annually. In addition, the FDA may, at any time during the 30-day period or at any time thereafter, impose a clinical hold on proposed or ongoing clinical trials. If the FDA imposes a clinical hold, clinical trials under the IND cannot commence or recommence without FDA authorization and then only under terms authorized by the FDA. An FDA imposed clinical hold on an IND application can result in substantial delay and large, unforeseen expenses, and it may cancel the viability of developing a new drug candidate in the United States.
Clinical trials are typically conducted in three sequential phases that may overlap. Phase I clinical trials typically study a drug’s safety profile, and may include the safe dosage range. Phase I clinical trials also determine how a drug is absorbed, distributed, metabolized and excreted by the body, and the duration of its action. In addition, we may, to the extent feasible, assess early indicators of a drug’s efficacy in our Phase I clinical trials. In Phase II clinical trials, controlled studies are conducted on volunteer patients with the targeted disease or condition. The primary purpose of these tests is to evaluate the effectiveness of the drug on the volunteer patients as well as to determine a drug’s side effect profile. These clinical trials may be conducted concurrently with Phase I clinical trials. In addition, Phase I/II clinical trials may be conducted to evaluate not only the efficacy of the drug on the patient population, but also its safety. During Phase III clinical trials, the drug is studied in an expanded patient population and in multiple sites. Physicians monitor the patients to determine efficacy and to observe and report adverse events that may result from use of the drug.
Our clinical trials are designed to produce clinical information about how our drugs perform compared to placebo or compared to existing opioid drugs where appropriate. We have designed most Phase II and Phase III clinical trials to date as randomized, double-blind, placebo-controlled, dose-ranging studies. A randomized clinical trial is one in which patients are randomly assigned to the various study treatment arms. A double-blind clinical trial is one in which the patient, the physician and our trial monitor are unaware if the patient is receiving placebo or study drug in order to preserve the integrity of the clinical trial and reduce bias. A placebo-controlled clinical trial is one in which a subset of patients is purposefully given inactive medication.
We may not successfully complete Phase I, Phase II or Phase III clinical trials within any specified time period, or at all, with respect to any of our drug candidates. Furthermore, we or the FDA may suspend clinical trials at any time in response to concerns that participants are exposed to an unacceptable health risk.
8
Table of Contents
After the completion of clinical trials, if there is substantial evidence that the drug is safe and effective, an NDA is filed with the FDA. The NDA must contain all of the information on the drug gathered to that date, including data from the clinical trials. NDAs are often the equivalent of over 100,000 pages in length.
The FDA reviews all NDAs submitted before it accepts them for filing and may request additional information rather than accepting an NDA for filing. In such an event, the NDA must be resubmitted with the additional information and, again, is subject to review before filing. Once the submission is accepted for filing, the FDA begins an in-depth review of the NDA. Under the Federal Food, Drug and Cosmetic Act, the FDA has 180 days in which to review the NDA and respond to the applicant. The review process is typically extended for significant amounts of time by the FDA’s requests for additional information or clarification regarding information already provided in the submission. The FDA may refer the application to an appropriate advisory committee, typically a panel of clinicians, for review, evaluation and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee. If the FDA’s evaluations of the NDA and the manufacturing facilities are favorable, the FDA may issue either a Complete Response Letter indicating either an approval or may identify conditions that must be met in order to secure final approval of the NDA. When and if those conditions have been met to the FDA’s satisfaction, the FDA will issue an approval letter, authorizing commercial marketing of the drug for certain indications. If the FDA’s evaluation of the NDA submission or manufacturing facilities is not favorable, the FDA may refuse to approve the NDA or issue a not approvable letter.
If the FDA approves the NDA, the drug becomes available for physicians to prescribe. Periodic reports must be submitted to the FDA, including descriptions of any adverse reactions reported. The FDA may request additional post marketing studies, or Phase IV clinical trials, to evaluate long-term effects of the approved drug.
The process for FDA approval of a BLA is similar to the process for FDA approval of an NDA.
Other Regulatory Requirements
The FDA mandates that drugs be manufactured in conformity with current GMP. If the FDA approves any of our drug candidates we will be subject to requirements for labeling, advertising, record keeping and adverse experience reporting. Failure to comply with these requirements could result, among other things, in suspension of regulatory approval, recalls, injunctions or civil or criminal sanctions. We may also be subject to regulations under other federal, state, and local laws, including the Occupational Safety and Health Act, the Environmental Protection Act, the Clean Air Act, national restrictions on technology transfer, and import, export, and customs regulations. In addition, any of our products that contain narcotics will be subject to DEA regulations relating to manufacturing, storage, distribution and physician prescribing procedures. It is possible that any portion of the regulatory framework under which we operate may change and that such change could have a negative impact on our current and anticipated operations.
The Controlled Substances Act imposes various registration, record-keeping and reporting requirements, procurement and manufacturing quotas, labeling and packaging requirements, security controls and a restriction on prescription refills on certain pharmaceutical products. A principal factor in determining the particular requirements, if any, applicable to a product is its actual or potential abuse profile. The DEA regulates chemical compounds as Schedule I, II, III, IV or V substances, with Schedule I substances considered to present the highest risk of substance abuse and Schedule V substances the lowest risk. Any of our drug candidates that contain a scheduled substance will be subject to regulation by the DEA.
Competition
Our success will depend, in part, upon our ability to achieve market share at the expense of existing and established and future products in the relevant target markets. Existing and future products, therapies, technological approaches or delivery systems will compete directly with our products. Competing products may provide greater therapeutic benefits for a specific indication, or may offer comparable performance at a lower cost. Companies that currently sell generic or proprietary opioid formulations include, but are not limited to,
9
Table of Contents
Roxane Laboratories, Purdue Pharma, Abbott Laboratories, Cephalon, Endo Pharmaceuticals, Teva Pharmaceuticals, Elkins-Sinn, Watson Laboratories, Ortho-McNeil Pharmaceutical and Forest Pharmaceuticals. Alternative technologies are being developed to increase opioid potency, as well as alternatives to opioid therapy for pain management, and improved treatments for metastatic melanoma and hemophilia, several of which are in clinical trials or are awaiting approval from the FDA.
We compete with fully integrated pharmaceutical companies, smaller companies that are collaborating with larger pharmaceutical companies, academic institutions, government agencies and other public and private research organizations. Many of these competitors have opioid drugs already approved by the FDA or in development and operate larger research and development programs in these fields than we do. In addition, many of these competitors, either alone or together with their collaborative partners, have substantially greater financial resources than we do, as well as significantly greater experience in:
| • | developing drugs; |
| • | undertaking preclinical testing and human clinical trials; |
| • | obtaining FDA and other regulatory approvals of drugs; |
| • | formulating and manufacturing drugs; and |
| • | launching, marketing, distributing and selling drugs. |
Developments by competitors may render our drug candidates or technologies obsolete or non-competitive. We also compete with these companies for qualified personnel and opportunities for product acquisitions, joint ventures or other strategic alliances
Incorporation
We were incorporated in Delaware in May 1998.
Employees
As of December 31, 2008, we had 48 employees. We engage consultants from time to time to perform services on a per diem or hourly basis.
Available Information
We file electronically with the Securities and Exchange Commission, or SEC, our annual reports on Form 10-K, quarterly reports on Form 10-Q and current reports on Form 8-K pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934. The public may read or copy any materials we file with the SEC at the SEC’s Public Reference Room at 450 Fifth Street, NW, Washington, DC 20549. The public may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC maintains an Internet site that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC. The address of that site is http://www.sec.gov.
You may obtain a free copy of our annual reports on Form 10-K, quarterly reports on Form 10-Q and current reports on Form 8-K and amendments to those reports on the day of filing with the SEC on our website on the World Wide Web at http://www.paintrials.com, by contacting the Investor Relations Department at our corporate offices by calling 650-624-8200 or by sending an e-mail message to cwaarich@paintrials.com.
| Item 1A. | Risk Factors |
Our future operating results may vary substantially from anticipated results due to a number of factors, many of which are beyond our control. The following discussion highlights some of these factors and the possible impact of these factors on future results of operations. You should carefully consider these factors
10
Table of Contents
before making an investment decision. If any of the following factors actually occur, our business, financial condition or results of operations could be harmed. In that case, the price of our common stock could decline, and you could experience losses on your investment in our common stock.
Clinical and Regulatory Risks
If we fail to obtain the necessary regulatory approvals, or if such approval is limited, we will not be allowed to commercialize our drug candidates, and we will not generate product revenues.
Satisfaction of all regulatory requirements typically takes many years, is dependent upon the type, complexity and novelty of the drug candidate, and requires the expenditure of substantial resources for research and development. In June 2008, we filed a New Drug Application, or NDA with the U.S. Food and Drug Administration, or FDA for our drug candidate Remoxy. In December 2008, we received a Complete Response Letter from the FDA for our NDA for Remoxy. The FDA indicated additional non-clinical data will be required to support the approval of REMOXY. We plan to meet with the FDA in the second quarter of 2009 regarding the NDA for Remoxy. We believe this FDA meeting will provide us with a more reliable context in which to make projections about Remoxy. If the FDA were to require additional data, providing such data may significantly delay the potential approval of Remoxy.
Our research and clinical approaches may not lead to drugs that the FDA considers safe for humans and effective for indicated uses we are studying. The FDA may require us to conduct additional non-clinical studies, in which case we would have to expend additional time and resources and would likely delay the date of potentially receiving regulatory approval. The approval process may also be delayed by changes in government regulation, future legislation or administrative action or changes in FDA policy that occur prior to or during our regulatory review. Delays in obtaining regulatory approvals would:
| • | delay commercialization of, and product revenues from, our drug candidates; and |
| • | diminish the competitive advantages that we may have otherwise enjoyed, which would have an adverse effect on our operating results and financial condition. |
Even if we comply with all FDA regulatory requirements, we may never obtain regulatory approval for any of our drug candidates. If we fail to obtain regulatory approval for any of our drug candidates we will have fewer commercial products, if any, and corresponding lower product revenues, if any. Even if we receive regulatory approval of our drug candidates, such approval may involve limitations on the indications and conditions of use or marketing claims we may make for our products. Further, later discovery of previously unknown problems or adverse events could result in additional regulatory restrictions, including withdrawal of products. The FDA may also require us to commit to perform lengthy Phase IV post-approval clinical trials, for which we would have to expend additional resources, which could have an adverse effect on our operating results and financial condition.
In jurisdictions outside the United States, we must receive marketing authorizations from the appropriate regulatory authorities before we can commercialize our drugs. Regulatory approval processes outside the United States generally include all of the aforementioned requirements and risks associated with FDA approval.
If we are unable to design, conduct and complete clinical trials successfully, we will not be able to obtain regulatory approval for our drug candidates.
In order to obtain FDA approval for any of our drug candidates, we must submit to the FDA an NDA that demonstrates with substantive evidence that the drug candidate is both safe and effective in humans for its intended use. This demonstration requires significant research and animal tests, which are referred to as preclinical studies, as well as human tests, which are referred to as clinical trials.
Results from our Phase I clinical programs may not support moving a drug candidate to Phase II or Phase III clinical trials. Our Phase III clinical trials may not demonstrate the safety or efficacy of our drug candidates.
11
Table of Contents
Success in preclinical studies and early clinical trials does not ensure that later clinical trials will be successful. Results of later clinical trials may not replicate the results of prior clinical trials and preclinical studies. Even if the results of our Phase III clinical trials are positive, we may have to commit substantial time and additional resources to conducting further preclinical studies and clinical trials before we can obtain FDA approval for any of our drug candidates.
Clinical trials are very expensive and difficult to design and implement, in part because they are subject to rigorous requirements. The clinical trial process also consumes a significant amount of time. Furthermore, if participating patients in clinical trials suffer drug-related adverse reactions during the course of such clinical trials, or if we or the FDA believe that participating patients are being exposed to unacceptable health risks, we will have to suspend or terminate our clinical trials. Failure can occur at any stage of the clinical trials, and we could encounter problems that cause us to abandon or repeat clinical trials.
Our clinical trials with Remoxy and our future clinical trials for other drug candidates for treatment of pain measure clinical symptoms, such as pain and physical dependence. These symptoms are not biologically measurable. The success of Remoxy and our other abuse resistant drug candidates in clinical trials depends on reaching statistically significant changes in patients’ symptoms based on clinician-rated scales. Due in part to a lack of consensus on standardized processes for assessing clinical outcomes, these scores may or may not be reliable, useful or acceptable to regulatory agencies.
We have no history of developing metastatic melanoma or hemophilia drug candidates. We do not know whether any of our planned clinical trials in metastatic melanoma or hemophilia will result in marketable drugs.
In addition, completion of clinical trials can be delayed by numerous factors, including:
| • | delays in identifying and agreeing on acceptable terms with prospective clinical trial sites; |
| • | slower than expected rates of patient recruitment and enrollment; |
| • | unanticipated patient drop out rates; |
| • | increases in time required to complete monitoring of patients during or after participation in a clinical trial; and |
| • | unexpected need for additional patient-related data. |
Any of these delays could significantly impact the timing, approval and commercialization of our drug candidates and could significantly increase our overall costs of drug development.
Even if our clinical trials are completed as planned, their results may not support our expectations or intended marketing claims. The clinical trials process may fail to demonstrate that our drug candidates are safe and effective for indicated uses. Such failure would cause us to abandon a drug candidate and could delay development of other drug candidates.
Clinical trial designs that were discussed with authorities prior to their commencement may subsequently be considered insufficient for approval at the time of application for regulatory approval.
We discuss with and obtain guidance from regulatory authorities on certain of our clinical development activities. With the exception of our Special Protocol Assessment, or SPA, such as the one we completed with the FDA with respect to our Phase III clinical trial for Remoxy, these discussions are not binding obligations on the part of regulatory authorities.
Regulatory authorities may revise previous guidance or decide to ignore previous guidance at any time during the course of our clinical activities or after the completion of our clinical trials. Even with successful clinical safety and efficacy data, including such data from a clinical trial conducted pursuant to an SPA, we may be required to conduct additional, expensive clinical trials to obtain regulatory approval.
12
Table of Contents
Developments by competitors may establish standards of care that affect our ability to conduct our clinical trials as planned.
We have conducted clinical trials of our drug candidates comparing our drug candidates to both placebo and other approved drugs. Changes in standards related to clinical trial design could affect our ability to design and conduct clinical trials as planned. For example, regulatory authorities may not allow us to compare our drug candidates to placebo in a particular clinical indication where approved products are available. In that case, both the cost and the amount of time required to conduct a clinical trial could increase.
The DEA limits the availability of the active ingredients in certain of our current drug candidates and, as a result, our quotas may not be sufficient to complete clinical trials, or to meet commercial demand or may result in clinical delays.
The U.S. Drug Enforcement Administration, or DEA, regulates chemical compounds as Schedule I, II, III, IV or V substances, with Schedule I substances considered to present the highest risk of substance abuse and Schedule V substances the lowest risk. Certain active ingredients in our current drug candidates, such as oxycodone, are listed by the DEA as Schedule II under the Controlled Substances Act of 1970. Consequently, their manufacture, research, shipment, storage, sale and use are subject to a high degree of oversight and regulation. For example, all Schedule II drug prescriptions must be signed by a physician, physically presented to a pharmacist and may not be refilled without a new prescription. Furthermore, the amount of Schedule II substances we can obtain for clinical trials and commercial distribution is limited by the DEA and our quota may not be sufficient to complete clinical trials or meet commercial demand. There is a risk that DEA regulations may interfere with the supply of the drugs used in our clinical trials, and, in the future, our ability to produce and distribute our products in the volume needed to meet commercial demand.
Conducting clinical trials of our drug candidates or potential commercial sales of a drug candidate may expose us to expensive product liability claims and we may not be able to maintain product liability insurance on reasonable terms or at all.
The risk of product liability is inherent in the testing of pharmaceutical products. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit or terminate testing of one or more of our drug candidates. Our inability to obtain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of our drug candidates. We currently carry clinical trial insurance but do not carry product liability insurance. If we successfully commercialize one or more of our drug candidates, we may face product liability claims, regardless of FDA approval for commercial manufacturing and sale. We may not be able to obtain such insurance at a reasonable cost, if at all. Even if our agreements with any current or future corporate collaborators entitle us to indemnification against product liability losses, such indemnification may not be available or adequate should any claim arise.
If we receive regulatory approval for our drug candidates, we and our collaborators will also be subject to ongoing FDA obligations and continued regulatory review, such as continued safety reporting requirements, and we and our collaborators may also be subject to additional FDA post-marketing obligations or new regulations, all of which may result in significant expense and limit our ability to commercialize our potential drugs.
Any regulatory approvals that we receive for our drug candidates may also be subject to limitations on the indicated uses for which the drug may be marketed or contain requirements for potentially costly post-marketing follow-up studies. In addition, if the FDA approves any of our drug candidates, the labeling, packaging, adverse event reporting, storage, advertising, promotion and record keeping for the drug will be subject to extensive regulatory requirements. The subsequent discovery of previously unknown problems with the drug, including but not limited to adverse events of unanticipated severity or frequency, or the discovery that adverse events
13
Table of Contents
previously observed in preclinical research or clinical trials that were believed to be minor actually constitute much more serious problems, may result in restrictions on the marketing of the drug, and could include withdrawal of the drug from the market.
The FDA’s policies may change and additional government regulations may be enacted that could prevent or delay regulatory approval of our drug candidates. For example, on February 9, 2009, the FDA announced it had invited the sponsors of opioid drug products to a private meeting in March 2009 in order to begin the process of developing Risk Evaluation and Mitigation Strategies for opioid drugs. According to FDA’s announcement, opioid drugs affected by this announcement include both brand name and generic products that contain fentanyl, hydromorphone, methadone, morphine, oxycodone and oxymorphone. We cannot predict the likelihood, nature or extent of adverse government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are not able to maintain regulatory compliance, we may be subject to fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution. Any of these events could prevent us from marketing our drugs and our business could suffer.
Risks Relating to Commercialization
If physicians and patients do not accept and use our drugs, we will not achieve sufficient product revenues and our business will suffer.
Even if the FDA approves our drugs, physicians and patients may not accept and use them. Acceptance and use of our drugs will depend on a number of factors including:
| • | perceptions by members of the healthcare community, including physicians, about the safety, effectiveness, and abuse resistant qualities of Remoxy; |
| • | perceptions by physicians regarding the cost benefit of the abuse resistant quality of Remoxy; |
| • | published studies demonstrating the cost-effectiveness of our drugs relative to competing products; |
| • | availability of reimbursement for our products from government or healthcare payers; |
| • | our ability to implement a risk management plan prior to the distribution of any Schedule II drug; and |
| • | effectiveness of marketing and distribution efforts by King, us and other licensees and distributors. |
Because we expect to rely on sales generated by our current lead drug candidates for substantially all of our revenues for the foreseeable future, the failure of any of these drugs to find market acceptance would harm our business and could require us to seek additional financing.
If King is not successful in commercializing Remoxy and other abuse resistant opioid drugs, our revenues and our business will suffer.
Our ability to earn royalties from sales of Remoxy and other abuse-resistant drugs will depend on King’s abilities to maintain regulatory approval and achieve market acceptance of such drugs once commercialized. King may elect to independently develop drugs that could compete with ours or fail to commit sufficient resources to the marketing and distribution of Remoxy and other abuse-resistant drugs developed under our strategic alliance. King may not proceed with the commercialization of Remoxy and other abuse-resistant drugs developed under our strategic alliance with the same degree of urgency as we would because of other priorities they face. If King is not successful in commercializing Remoxy for a variety of reasons, including but not limited to competition from other pharmaceutical companies, or if King fails to perform as we expect, our potential for revenue from drugs developed in connection with our strategic alliance with King, if any, could be dramatically reduced and our business would suffer.
14
Table of Contents
If we are unable to develop our own sales, marketing and distribution capabilities, or if we are not successful in contracting with third parties for these services on favorable terms, or at all, our product revenues could be disappointing.
We currently have no sales, marketing or distribution capabilities. Except with regard to products developed under our strategic alliance with King, in order to commercialize our products, if any are approved by the FDA, we will either have to develop such capabilities internally or collaborate with third parties who can perform these services for us. If we decide to commercialize any of our drugs ourselves, we may not be able to hire the necessary experienced personnel and build sales, marketing and distribution operations which are capable of successfully launching new drugs and generating sufficient product revenues. In addition, establishing such operations will take time and involve significant expense.
If we decide to enter into new co-promotion or other licensing arrangements with third parties, we may be unable to locate acceptable collaborators because the number of potential collaborators is limited and because of competition from others for similar alliances with potential collaborators. Even if we are able to identify one or more acceptable new collaborators, we may not be able to enter into any collaborative arrangements on favorable terms, or at all.
In addition, due to the nature of the market for our drug candidates, it may be necessary for us to license all or substantially all of our drug candidates not covered by our strategic alliance with King to a single collaborator, thereby eliminating our opportunity to commercialize these other products independently. If we enter into any such new collaborative arrangements, our revenues are likely to be lower than if we marketed and sold our products ourselves.
In addition, any revenues we receive would depend upon our collaborators’ efforts which may not be adequate due to lack of attention or resource commitments, management turnover, change of strategic focus, business combinations or other factors outside of our control. Depending upon the terms of our collaboration, the remedies we have against an under-performing collaborator may be limited. If we were to terminate the relationship, it may be difficult or impossible to find a replacement collaborator on acceptable terms, or at all.
If we cannot compete successfully for market share against other drug companies, we may not achieve sufficient product revenues and our business will suffer.
The market for our drug candidates is characterized by intense competition and rapid technological advances. If our drug candidates receive FDA approval, they will compete with a number of existing and future drugs and therapies developed, manufactured and marketed by others. Existing or future competing products may provide greater therapeutic convenience or clinical or other benefits for a specific indication than our products, or may offer comparable performance at a lower cost. If our products are unable to capture and maintain market share, we may not achieve sufficient product revenues and our business will suffer.
We and our collaborators will compete for market share against fully integrated pharmaceutical companies or other companies that are collaborating with larger pharmaceutical companies, academic institutions, government agencies and other public and private research organizations. Many of these competitors have drugs already approved or drug candidates in development that will or may compete against our approved drug candidates. In addition, many of these competitors, either alone or together with their collaborative partners, operate larger research and development programs and have substantially greater financial resources than we do, as well as significantly greater experience in:
| • | developing drugs; |
| • | conducting preclinical testing and human clinical trials; |
| • | obtaining FDA and other regulatory approvals of drugs; |
| • | formulating and manufacturing drugs; and |
15
Table of Contents
| • | launching, marketing, distributing and selling drugs. |
Our ability to generate product revenues will be diminished if we fail to obtain acceptable prices or an adequate level of reimbursement for our products from healthcare payers.
Our ability to commercialize our drugs, alone or with collaborators, will depend in part on the extent to which reimbursement will be available from:
| • | government and health administration authorities; |
| • | private health maintenance organizations and health insurers; and |
| • | other healthcare payers. |
Significant uncertainty exists as to the reimbursement status of newly approved healthcare products. Healthcare payers, including Medicare, health maintenance organizations and managed care organizations, are challenging the prices charged for medical products and services and/or are seeking pharmacoeconomic data to justify formulary acceptance and reimbursement practices. We currently have not generated pharmacoeconomic data on any of our drug candidates. Government and other healthcare payers increasingly are attempting to contain healthcare costs by limiting both coverage and the level of reimbursement for drugs, and by refusing, in some cases, to provide coverage for uses of approved products for disease indications for which the FDA has or has not granted labeling approval. Adequate third-party insurance coverage may not be available to patients for any products we discover and develop, alone or with collaborators. If government and other healthcare payers do not provide adequate coverage and reimbursement levels for our products, market acceptance of our drug candidates could be limited.
Government agencies may establish and promulgate usage guidelines that could limit the use of our drug candidates.
Government agencies, professional and medical societies, and other groups may establish usage guidelines that apply to our drug candidates. These guidelines could address such matters as usage and dose, among other factors. Application of such guidelines could limit the clinical use or commercial appeal of our drug candidates.
Risks Relating to our Collaboration Agreements
If King or other outside collaborators fail to devote sufficient time and resources to our drug development programs, or if their performance is substandard, our regulatory submissions and our product introductions may be delayed.
Pursuant to our strategic alliance with King, we jointly manage and prepare Phase III clinical trials and NDA submissions in the United States for Remoxy and other abuse-resistant drug candidates with King. We rely on King to devote time and resources to the development, manufacturing and commercialization of Remoxy and other abuse-resistant drug candidates. King may develop or acquire drugs or drug candidates that compete for resources with our drug candidates that are subject to this strategic alliance. For instance, King has acquired a drug candidate for the acute pain market and has acquired another company that has abuse resistant technologies and two NDAs in place with the FDA for abuse resistant opioids. While we believe these drug candidates will not compete directly with our drug candidates being developed under the collaboration with King, there can be no assurance that King’s other drug candidates will not become competitive with our drug candidates being developed under the collaboration with King. If King limits its time and resources devoted to the strategic alliance or fails to fund the continued development of PTI-202, PTI-721, or other abuse-resistant opioid potential products as required by our agreement with King, or otherwise fails to perform as we expect, we may not achieve clinical and regulatory milestones and regulatory submissions and related product introductions may be delayed or prevented, and revenues that we would receive from these activities will be less than expected. In addition, if King fails to perform as required under our collaboration agreement, their failure may jeopardize our rights under our license with Durect.
16
Table of Contents
We depend on independent investigators and collaborators, such as universities and medical institutions, to conduct our clinical trials under agreements with us. These investigators and collaborators are not our employees and we cannot control the amount or timing of resources that they devote to our programs. They may not assign as great a priority to our programs or pursue them as diligently as we would if we were undertaking such activities ourselves. If these investigators or collaborators fail to devote sufficient time and resources to our drug development programs, or if their performance is substandard, the approval of our regulatory submissions and our introductions of new drugs will be delayed or prevented.
Our collaborators may also have relationships with other commercial entities, some of which may compete with us. If outside collaborators assist our competitors to our detriment, the approval of our regulatory submissions will be delayed and the sales from our products, if any are commercialized, will be less than expected.
If we fail to maintain our collaboration agreements and licenses for Remoxy and other abuse-resistant drugs, we may have to reduce or delay our drug candidate development.
Our plan for developing, manufacturing and commercializing Remoxy and other abuse-resistant drugs currently requires us to successfully maintain our strategic alliance with King to advance our programs and provide funding to support our expenditures on Remoxy and other drug candidates and to maintain our license from Durect. If we are not able to maintain our existing strategic alliance with King or if King doesn’t provide the required funding under the strategic alliance, we may have to limit the size or scope of, or delay or abandon the development of Remoxy and other abuse-resistant drug candidates or undertake and fund development of these drug candidates ourselves and if we are unable to maintain our license with Durect for one or more potential products we may lose the rights to utilize Durect’s technology for such potential products. If we elect to fund drug development efforts with respect to Remoxy and other abuse-resistant drug candidates on our own, we may need to obtain additional capital, which may not be available on acceptable terms, or at all.
We may not succeed at in-licensing drug candidates or technologies to expand our product pipeline.
We may not successfully in-license drug candidates or technologies to expand our product pipeline. The number of such candidates or technologies is limited. Competition among large pharmaceutical companies and biopharmaceutical companies for promising drug candidates or technologies is intense because such companies generally desire to expand their product pipelines through in-licensing. If we fail to carry out such in-licensing and expand our product pipeline, our potential future revenues may suffer.
Our collaborative agreements may not succeed or may give rise to disputes over intellectual property, disputes concerning the scope of collaboration activities or other issues.
Our strategy to focus on drug development requires us to enter into collaborative agreements with third parties, such as our strategic alliance with King and our license agreement with Durect. Such agreements are generally complex and contain provisions that could give rise to legal disputes, including potential disputes concerning ownership of intellectual property under collaborations or disputes concerning the scope of collaboration activities. Such disputes can delay or prevent the development of potential new drug products, or can lead to lengthy, expensive litigation or arbitration. Other factors relating to collaborative agreements may adversely affect the success of our drug candidates, including:
| • | the development of parallel products by our collaborators or by a competitor; |
| • | arrangements with collaborative partners that limit or preclude us from developing certain products or technologies; |
| • | premature termination of a collaborative or license agreement; or |
| • | failure by a collaborative partner to provide required funding or to devote sufficient resources to the development of or legal defense of our potential products. |
17
Table of Contents
Risks Relating to our Intellectual Property
Our ability to commercialize our drug candidates will depend on our ability to sell such products without infringing the patent or proprietary rights of third parties. If we are sued for infringing the intellectual property rights of third parties, such litigation will be costly and time consuming and an unfavorable outcome would have a significant adverse effect on our business.
Our ability to commercialize our drug candidates will depend on our ability to sell such products without infringing the patents or other proprietary rights of third parties. Intellectual property rights in the areas of controlled-release oxycodone, antibodies, gene integration and more generally, in oncology, neurology, radiopharmaceutical technologies and gene therapy are complicated and are continuously evolving. Holders of patent rights in these areas may allege that the commercialization of Remoxy or our other drug candidates infringes such patent rights. While we believe that we would have valid defenses to any claim of infringement, there can be no assurance that these or other third party patents will not limit our ability to commercialize Remoxy or our other drug candidates.
In addition, because patent applications are published 18 months after their filing, and because applications can take several years to issue, there may be currently pending third-party patent applications that are unknown to us, which may later result in issued patents. If a third-party claims that we infringe on its patents or other proprietary rights, we could face a number of issues that could seriously harm our competitive position, including:
| • | infringement claims that, with or without merit, can be costly and time consuming to litigate, can delay the regulatory approval process and can divert management’s attention from our core business strategy; |
| • | substantial damages for past infringement which we may have to pay if a court determines that our products or technologies infringe upon a competitor’s patent or other proprietary rights; |
| • | a court order prohibiting us from commercializing our products or technologies unless the holder licenses the patent or other proprietary rights to us, which such holder is not required to do; |
| • | if a license is available from a holder, we may have to pay substantial royalties or grant cross licenses to our patents or other proprietary rights; and |
| • | redesigning our process so that it does not infringe the third-party intellectual property rights, which may not be possible, or which may require substantial time and expense including delays in bringing our own products to market. Such actions could harm our competitive position and our ability to generate revenue and could result in increased costs. |
If we are unable to protect our intellectual property our competitors could develop and market products with similar features that may reduce demand for our drug candidates.
Our success, competitive position and potential future revenues will depend in part on our ability to protect our intellectual property. If we or our collaborators fail to file, prosecute, obtain or maintain certain patents, our competitors could market products that contain features and clinical benefits similar to those of our products, and demand for our products could decline as a result.
We and our collaborators have filed patent applications with the U.S. Patent and Trademark Office to further protect our technologies. If these patent applications do not result in issued patents, the duration or scope of our patent rights may be limited and our future revenues could be lower as a result.
We may be involved in challenges to our intellectual property. An adverse outcome of a challenge to our intellectual property could result in loss of claims of patents or other intellectual property rights that pertain to certain drugs we currently have under development and could have a material adverse impact on our future revenues.
18
Table of Contents
We intend to file additional patent applications relating to our technology, products and processes. We may direct our collaborators to file additional patent applications relating to the licensed technology or we may do so ourselves. However, our competitors may challenge, invalidate or circumvent any of our current or future patents. These patents may also fail to provide us with meaningful competitive advantages.
We may become involved in expensive litigation or other legal proceedings related to our existing intellectual property rights, including patents.
We expect that we will rely upon patents, trade secrets, know-how, continuing technological innovations and licensing opportunities to develop and maintain our competitive position. Others may independently develop substantially equivalent proprietary information or be issued patents that may prevent the sale of our products or know-how or require us to license such information and pay significant fees or royalties in order to produce our products.
Our technology could infringe upon claims of patents owned by others. If we were found to be infringing on a patent held by another, we might have to seek a license to use the patented technology. In that case, we might not be able to obtain such a license on terms acceptable to us, or at all. If a legal action were to be brought against us or our licensors, we could incur substantial defense costs, and any such action might not be resolved in our favor. If such a dispute were to be resolved against us, we could have to pay the other party large sums of money and our use of our technology and the testing, manufacture, marketing or sale of one or more of our proposed products could be restricted or prohibited.
Risks Relating to our Business and Strategy
Competition for qualified personnel in the pharmaceutical industry is intense, and if we are not successful in attracting and retaining qualified personnel, we could experience delays in completing necessary clinical trials, in the regulatory approval process or in formulating, manufacturing, marketing and selling our potential products.
We will need to hire additional qualified personnel with expertise in clinical research, preclinical testing, government regulation, formulation and manufacturing and sales and marketing. We compete for qualified individuals with numerous biopharmaceutical companies, universities and other research institutions. Competition for such individuals, particularly in the San Francisco Bay area, is intense, and our search for such personnel may not be successful. Attracting and retaining qualified personnel is critical to our success.
If third-party manufacturers of our drug candidates fail to devote sufficient time and resources to our concerns, or if their performance is substandard, our clinical trials and product introductions may be delayed and our costs may be higher than expected.
We have no manufacturing facilities and have limited experience in drug product development and commercial manufacturing. We lack the resources and expertise to formulate, manufacture or test the technical performance of our drug candidates. We currently rely on a limited number of experienced personnel and a small number of contract manufacturers and other vendors to formulate, test, supply, store and distribute drug supplies for our clinical trials. Our reliance on a limited number of vendors exposes us to the following risks, any of which could delay our clinical trials, and, consequently, FDA approval of our drug candidates and commercialization of our products, result in higher costs, or deprive us of potential product revenues:
| • | Contract commercial manufacturers, their sub-contractors or other third parties we rely on, may encounter difficulties in achieving the volume of production needed to satisfy clinical needs or commercial demand, may experience technical issues that impact quality or compliance with applicable and strictly enforced regulations governing the manufacture of pharmaceutical products, and may experience shortages of qualified personnel to adequately staff production operations. |
| • | Our contract manufacturers could default on their agreements with us to provide clinical supplies or meet our requirements for commercialization of our products. |
19
Table of Contents
| • | For certain of our drug candidates, the use of alternate manufacturers may be difficult because the number of potential manufacturers that have the necessary governmental licenses to produce narcotic products is limited. Additionally, the FDA and the DEA must approve any alternative manufacturer of our products before we may use the alternative manufacturer to produce our supplies. |
| • | It may be difficult or impossible for us to find a replacement manufacturer on acceptable terms quickly, or at all. Our contract manufacturers and vendors may not perform as agreed or may not remain in the contract manufacturing business for the time required to successfully produce, store and distribute our products. |
| • | If any contract manufacturer makes improvements in the manufacturing process for our products, we may not own, or may have to share, the intellectual property rights to such innovation. |
We expanded our research and development activities to include development of potential drug candidates for indications other than pain, and we may not be able to successfully develop or commercialize these potential new drug candidates.
We have expanded our research and development activities to include development of potential drug candidates for indications other than pain, such as metastatic melanoma and hemophilia. We completed a Phase I clinical trial of our drug candidate in metastatic melanoma in 2008, and plan to initiate another study in such indication. We have no history of developing metastatic melanoma or hemophilia drug candidates or manufacturing radiopharmaceuticals. We do not know whether any of our planned clinical trials in metastatic melanoma or hemophilia will result in marketable products. We do not anticipate that any drug candidates in these areas will reach the market for at least several years, if at all.
Our employees and consultants are generally subject to confidentiality or other agreements with their former employers and they may inadvertently or otherwise violate those agreements.
Many of our employees and consultants were previously employed at universities or biotechnology or pharmaceutical companies. While we require our employees and consultants to honor any agreements they may have entered into prior to working with us, we may be subject to claims that we inadvertently or otherwise used or disclosed trade secrets or other confidential information belonging to former employers. Failure to defend such claims could result in loss of valuable rights or personnel, which in turn could harm or prevent commercialization of our drug candidates. Successful defense against such claims can be expensive and might distract us from executing our strategies.
Law enforcement concerns over diversion of opioids and social issues around abuse of opioids may make the regulatory approval process and commercialization of our drug candidates very difficult.
Media stories regarding the diversion of opioids and other controlled substances are commonplace. Law enforcement agencies or regulatory agencies may apply policies that seek to limit the availability of opioids. Such efforts may adversely affect the regulatory approval and commercialization of our drug candidates.
Developments by competitors may render our products or technologies obsolete or non-competitive.
Alternative technologies and products are being developed to improve or replace the use of opioids for pain management, several of which are in clinical trials or are awaiting approval from the FDA. In addition, the active ingredients in nearly all opioid drugs are available in generic form. Drug companies that sell generic opioid drugs represent substantial competition. Many of these organizations competing with us have substantially greater capital resources, larger research and development staffs and facilities, greater experience in drug development and in obtaining regulatory approvals and greater manufacturing and marketing capabilities than we do. Our competitors may market less expensive or more effective drugs that would compete with our drug candidates or reach market with competing drugs before we are able to reach market with our drug candidates. These organizations also compete with us to attract qualified personnel and partners for acquisitions, joint ventures or other collaborations.
20
Table of Contents
Business interruptions could limit our ability to operate our business.
Our operations as well as those of our collaborators on which we depend are vulnerable to damage or interruption from computer viruses, human error, natural disasters, electrical and telecommunication failures, international acts of terror and similar events. We have not established a formal disaster recovery plan and our back-up operations and our business interruption insurance may not be adequate to compensate us for losses we may suffer. A significant business interruption could result in losses or damages incurred by us and require us to cease or curtail our operations.
Risks Relating to Manufacturing
We rely on third-party commercial drug manufacturers for drug supply.
Approved third-party commercial drug manufacturers may subsequently be stopped from producing, storing, shipping or testing our drug products due to their non-compliance with federal, state or local regulations. Drug manufacturers are subject to ongoing periodic unannounced inspection by the FDA, the DEA, and corresponding state and foreign government agencies to ensure strict compliance with GMP and other government regulations and corresponding foreign standards. We do not have control over third-party manufacturers’ compliance with these regulations and standards.
In addition, even if we enter into long-term supply arrangements with third-party suppliers, we cannot control changes in strategy by third-party suppliers that affect their ability or willingness to continue to supply our drug products on acceptable terms.
If our drug supply for one of our drug candidates was interrupted, our operations could be negatively affected.
If we cannot formulate and scale-up a wide range of dosage forms of Remoxy and other abuse-resistant drug candidates, we and King might determine that the commercial opportunity for Remoxy and other abuse resistant drug candidates in certain dosage forms is too limited to warrant further investment in clinical testing and development.
We plan to formulate and scale-up a wide range of dosage forms of Remoxy and other abuse-resistant drug candidates. We may not be able to successfully complete our formulation or scale-up activities or we may determine that the commercial opportunity for Remoxy and other abuse resistant drug candidates in certain dosage forms is too limited to warrant further investment. If we are unsuccessful in our formulation or scale-up activities with Remoxy and other abuse-resistant drug candidates, our future revenue from milestones and royalties under our strategic alliance with King may be less than expected and our operations may suffer.
We and King rely solely on Durect to provide us with certain components of Remoxy and other abuse-resistant drug candidates and will continue to rely on Durect to produce commercial supplies of these components.
We and King rely on Durect as the sole source provider of certain components of Remoxy and other abuse-resistant drug candidates, and will rely solely on Durect to produce commercial supplies of these components. Durect’s failure to achieve and maintain satisfactory manufacturing standards could result in product recalls or withdrawals, delays or failures in product testing or delivery, cost overruns or other problems that could materially harm our business. Durect may encounter manufacturing difficulties involving production yields, quality control and quality assurance. Durect is subject to ongoing periodic unannounced inspection by the FDA and corresponding state and foreign agencies to ensure strict compliance with government regulations and corresponding foreign standards. We cannot control Durect’s compliance with these regulations and standards.
To date, Durect has not produced commercial-scale supply of these components. If we and King receive marketing approval for and commercially launch Remoxy or other abuse resistant candidates, we anticipate that
21
Table of Contents
Durect will need to materially expand its manufacturing capacity. Durect may not be able to increase its manufacturing capacity for Remoxy and other abuse-resistant drug candidates in a timely or economic manner, or at all. Moreover, significant scale up of manufacturing will require additional validation studies, which are subject to FDA review and approval. If Durect is unable to successfully increase the manufacturing capacity for such components of Remoxy and other abuse-resistant drugs, at an acceptable cost or otherwise, and we are unable to establish alternative manufacturing capabilities, the commercial launch or continued commercialization after a commercial launch of Remoxy and other abuse-resistant drugs may be delayed, prevented or impaired or there may be a shortage in supply, which would harm our future revenues and cause our business to suffer.
Risks Relating to our Financial Position and Need for Financing
Our operating history may make it difficult for you to evaluate our business to date and to assess its future viability.
Our operations from our inception to date have been limited to organizing and staffing our company, acquiring, developing and securing our technology, undertaking preclinical studies and clinical trials of our drug candidates and forming collaborations. We have not yet demonstrated our ability to obtain regulatory approval, formulate and manufacture our drug candidates on a commercial scale or conduct sales and marketing activities. Consequently, any predictions you make about our future success or viability may not be as accurate as they could be if we had a longer operating history.
We have a history of losses and expect to incur substantial losses and negative operating cash flows for the foreseeable future.
Although we were profitable in 2006, 2007 and 2008 based on payments from King and interest income, we have yet to generate any revenues from product sales. We had an accumulated deficit of $114.1 million as of December 31, 2008. Even if we succeed in developing and commercializing one or more of our drug candidates, we expect to continue to use significant cash resources in our operations for the foreseeable future. We anticipate that our expenses will increase substantially in the foreseeable future as we:
| • | continue to conduct preclinical studies and clinical trials for our drug candidates; |
| • | seek regulatory approvals for our drug candidates; |
| • | develop, formulate, manufacture and commercialize our drug candidates; |
| • | implement additional internal systems and develop new infrastructure; |
| • | acquire or in-license additional products or technologies, or expand the use of our technology; |
| • | maintain, defend and expand the scope of our intellectual property; and |
| • | hire additional personnel. |
We will need to generate significant revenues to achieve and maintain profitability. If we cannot successfully develop, obtain regulatory approval for and commercialize our drug candidates, we will not be able to generate such revenues or achieve profitability in the future. Our failure to achieve or maintain profitability would have a material adverse impact on the market price of our common stock.
If we cannot raise additional capital on acceptable terms, we may be unable to complete planned clinical trials of any or some of our drug candidates or to pursue attractive business opportunities.
We have funded all of our operations and capital expenditures with the proceeds from our public and private stock offerings, payments received under our strategic alliance with King, and interest earned on our investments. We expect that our current cash, cash equivalents and marketable securities will be sufficient to meet our working capital and capital expenditure needs for at least the next twelve months. However, we may elect to raise additional funds within such twelve-month period or need to raise additional funds thereafter and additional
22
Table of Contents
financing may not be available on favorable terms, if at all. Even if we succeed in selling additional securities to raise funds, our existing stockholders’ ownership percentage would be reduced and new investors may demand rights, preferences or privileges senior to those of existing stockholders. If we raise additional capital through debt financing, if available, such financings may involve covenants that restrict our business activities. If we raise additional capital through strategic alliance and license arrangements such as our strategic alliance with King, we may have to trade our rights to our technology, intellectual property or drug candidates to others in such arrangements on terms that may not be favorable to us.
If we determine that we need to raise additional funds and we are not successful in doing so, we may be unable to complete the clinical development of some or all of our drug candidates or to seek or obtain FDA approval of our drug candidates. We then could be forced to discontinue product development, enter into a relationship with an additional strategic partner earlier than currently intended, reduce sales and marketing efforts or forego attractive business opportunities.
Risks Relating to an Investment in our Common Stock
Our stock price has been volatile and could experience a sudden decline in value.
Our common stock has experienced significant price and volume fluctuations and may continue to experience volatility in the future. You may not be able to sell your shares quickly or at the latest market price if trading in our stock is not active or the volume is low. The following factors, in addition to other risk factors described in this section, may have a significant impact on the market price of our common stock:
| • | results of or delays in our efforts to seek regulatory approval for Remoxy, and in preclinical studies and clinical trials for our other drug candidates; |
| • | the success of our collaboration agreements; |
| • | publicity regarding actual or potential medical results relating to products under development by us or others; |
| • | announcements of technological innovations or new commercial products by us or others; |
| • | developments in patent or other proprietary rights by us or others; |
| • | comments or opinions by securities analysts or major stockholders; |
| • | future sales of our common stock by existing stockholders; |
| • | regulatory developments or changes in regulatory guidance enacted by applicable governmental or other authorities; |
| • | litigation or threats of litigation; |
| • | economic and other external factors or other disaster or crises; |
| • | the departure of any of our officers, directors or key employees; |
| • | period-to-period fluctuations in financial results; and |
| • | limited daily trading volume. |
Changing laws, regulations and standards relating to corporate governance and public disclosure, including the Sarbanes-Oxley Act of 2002, SEC regulations and the rules of The NASDAQ Stock Market LLC create uncertainty for public companies. If we were unable to continue to comply with these requirements, we could be delisted from trading on the NASDAQ Global Market, or Nasdaq, and thereafter trading in our common stock, if any, may be conducted through the over-the-counter or other market. As a consequence of such delisting, an investor would likely find it more difficult to dispose of, or to obtain quotations as to the price of, our common stock. Delisting of our common stock could also result in lower prices per share of our common stock than would otherwise prevail.
23
Table of Contents
There may be “naked” short selling in our common stock, which may lead to extraordinary volatility in the market price of our common stock, even in the absence of new fundamental information regarding our business.
Short selling is a form of speculation that allows an investor to sell stock that they do not own. In order to short sell a stock, an investor normally borrows shares from a third party and agrees to return these shares at some future date. “Naked” short selling occurs when a trader short sells a stock with no actual delivery of shares taking place to close the transaction. The legality of this practice is unclear. As of January 15, 2009, Nasdaq reported short interest of approximately 4.9 million shares in our common stock. We believe this amount may represent a significant percentage of our outstanding common stock available to borrow. Such short interest may also include a significant amount of naked short selling. Widespread naked short selling may increase volatility of our common stock and may have a significant, transient and highly negative impact on the market price of our common stock.
Anti-takeover provisions in our charter documents, our Stockholder Rights Plan and Delaware law may prevent or delay removal of incumbent management or a change of control.
Anti-takeover provisions of our amended and restated certificate of incorporation and amended and restated bylaws, our Stockholder Rights Plan and Delaware law may have the effect of deterring or delaying attempts by our stockholders to remove or replace management, engage in proxy contests and effect changes in control. The provisions of our charter documents include:
| • | a classified board so that only one of the three classes of directors on our board of directors is elected each year; |
| • | elimination of cumulative voting in the election of directors; |
| • | procedures for advance notification of stockholder nominations and proposals; |
| • | the ability of our board of directors to amend our bylaws without stockholder approval; and |
| • | the ability of our board of directors to issue up to 10,000,000 shares of preferred stock without stockholder approval upon the terms and conditions and with the rights, privileges and preferences as our board of directors may determine. |
The rights issued pursuant to our Stockholder Rights Plan will become exercisable, subject to certain exceptions, the tenth day after a person or group announces acquisition of 15% or more of our common stock or announces commencement of a tender or exchange offer the consummation of which would result in ownership by the person or group of 15% or more of our common stock.
In addition, as a Delaware corporation, we are subject to Delaware law, including Section 203 of the Delaware General Corporation Law. In general, Section 203 prohibits a Delaware corporation from engaging in any business combination with any interested stockholder for a period of three years following the date that the stockholder became an interested stockholder unless certain specific requirements are met as set forth in Section 203.
These provisions, alone or together, could have the effect of deterring or delaying changes in incumbent management, proxy contests or changes in control.
Volatility in the stock prices of other companies may contribute to volatility in our stock price.
The stock market in general, Nasdaq and the market for technology companies in particular, have experienced significant price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of those companies. Further, there has been particular volatility in the market prices of securities of early stage life sciences companies. These broad market and industry factors may seriously harm the
24
Table of Contents
market price of our common stock, regardless of our operating performance. In the past, following periods of volatility in the market price of a company’s securities, securities class action litigation has often been instituted. A securities class action suit against us could result in substantial costs, potential liabilities and the diversion of management’s attention and resources.
Our share ownership is concentrated, and our officers, directors and principal stockholders can exert significant control over matters requiring stockholder approval.
Due to their combined stock holdings, our officers, directors and principal stockholders (stockholders holding greater than 5% of our common stock) acting collectively may have the ability to exercise significant influence over matters requiring stockholder approval including the election of directors and approval of significant corporate transactions. This concentration of ownership may delay or prevent a change in control of the Company and may make some transactions more difficult or impossible to complete without the support of these stockholders.
Publicly available information regarding stockholders’ ownership may not be comprehensive because the SEC does not require certain large stockholders to publicly disclose their stock ownership positions.
Our operating results may fluctuate from quarter to quarter and this fluctuation may cause our stock price to decline.
Our quarterly operating results have fluctuated in the past and are likely to fluctuate in the future. Factors contributing to these fluctuations include, among other items, the timing and amounts of collaboration revenue recognized from King, the timing and enrollment rates of clinical trials for our drug candidates, our need for clinical supplies and the valuation of stock-based compensation. Thus, quarter-to-quarter comparisons of our operating results are not indicative of what we might expect in the future. As a result, in some future quarters our clinical, financial or operating results may not meet the expectations of securities analysts and investors that could result in a decline in the price of our stock.
There may not be an active, liquid trading market for our common stock.
There is no guarantee that an active trading market for our common stock will be maintained on Nasdaq. Investors may not be able to sell their shares quickly or at the latest market price if trading in our stock is not active.
| Item 1B. | Unresolved Staff Comments |
None.
| Item 2. | Properties |
We currently lease approximately 41,200 square feet of space in San Mateo and South San Francisco, California. All of our operations are located at our office space in San Mateo. We believe that our facilities are adequate and suitable for our current needs.
| Item 3. | Legal Proceedings |
We are not a party to any legal proceedings.
| Item 4. | Submission of Matters to a Vote of Security Holders |
There were no matters submitted to a vote of the security holders during the fourth quarter of 2008.
25
Table of Contents
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Our common stock is quoted on the NASDAQ Global Market under the symbol “PTIE.” The following table sets forth the high and low sales prices per share of our common stock as reported on the NASDAQ Global Market for the periods indicated.
| Sales Prices | ||||||
| High | Low | |||||
| Fiscal 2007: |
||||||
| First Quarter |
$ | 9.20 | $ | 7.00 | ||
| Second Quarter |
$ | 9.00 | $ | 7.54 | ||
| Third Quarter |
$ | 9.86 | $ | 8.20 | ||
| Fourth Quarter |
$ | 11.50 | $ | 9.35 | ||
| Fiscal 2008: |
||||||
| First Quarter |
$ | 10.73 | $ | 7.48 | ||
| Second Quarter |
$ | 8.86 | $ | 6.80 | ||
| Third Quarter |
$ | 10.32 | $ | 7.35 | ||
| Fourth Quarter |
$ | 10.09 | $ | 5.50 | ||
We currently expect to retain future earnings, if any, for use in the operation and expansion of our business and have not paid and do not anticipate paying any cash dividends in the foreseeable future. As of January 12, 2009, there were approximately 72 holders of record of our common stock.
26
Table of Contents
Performance Graph
The following line graph compares the percentage change in the cumulative return to the stockholders of the our common stock with the cumulative return of the NASDAQ Composite Index and the NASDAQ Pharmaceutical Index for the period commencing December 31, 2003.
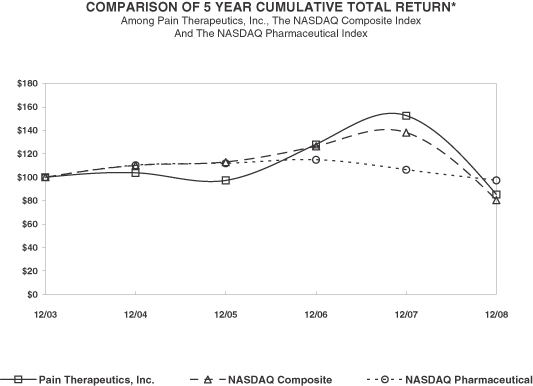
| * | The graph assumes that $100 was invested on 12/31/03 in our common stock or index and that all dividends were reinvested. We have not declared or paid any dividends on our common stock. Stockholder returns over the indicated period should not be considered indicative of future stockholder returns. |
27
Table of Contents
| Item 6. | Selected Financial Data (in thousands except per share data) |
The following selected financial data should be read together with the “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our financial statements and related notes included elsewhere in this report. The selected balance sheet data at December 31, 2008 and 2007 and the selected statement of operations data for each year ended December 31, 2008, 2007, and 2006 have been derived from our audited financial statements that are included elsewhere in this report. The selected balance sheets data at December 31, 2006, 2005, and 2004 and the statements of operations for each year ended December 31, 2005 and 2004 have been derived from our audited financial statements not included in this report. Historical results are not necessarily indicative of the results to be expected in the future.
| Years ended December 31, | |||||||||||||||||||
| 2008 (1) | 2007 (1) | 2006 (1) | 2005 | 2004 | |||||||||||||||
| Statement of operations data: |
|||||||||||||||||||
| Collaboration revenue |
$ | 29,377 | $ | 42,746 | $ | 22,717 | $ | 1,368 | $ | — | |||||||||
| Program fee revenue |
14,348 | 23,238 | 26,201 | 3,712 | — | ||||||||||||||
| Milestone revenue |
20,000 | — | 5,000 | — | — | ||||||||||||||
| Total revenue |
63,725 | 65,984 | 53,918 | 5,080 | — | ||||||||||||||
| Research and development expense |
45,817 | 47,730 | 46,803 | 32,938 | 35,093 | ||||||||||||||
| General and administrative expense |
9,196 | 8,085 | 7,668 | 4,859 | 3,868 | ||||||||||||||
| Total operating expenses |
55,013 | 55,815 | 54,471 | 37,797 | 38,961 | ||||||||||||||
| Operating income (loss) |
8,712 | 10,169 | (553 | ) | (32,717 | ) | (38,961 | ) | |||||||||||
| Interest and other income, net |
6,018 | 10,136 | 9,668 | 2,047 | 1,185 | ||||||||||||||
| Income (loss) before provision for (benefit from) income taxes |
14,730 | 20,305 | 9,115 | (30,670 | ) | (37,776 | ) | ||||||||||||
| Provision for (benefit from) income taxes |
(617 | ) | — | 2,927 | — | — | |||||||||||||
| Net income (loss) |
$ | 15,347 | $ | 20,305 | $ | 6,188 | $ | (30,670 | ) | $ | (37,776 | ) | |||||||
| Net income (loss) per share: |
|||||||||||||||||||
| Basic |
$ | 0.36 | $ | 0.46 | $ | 0.14 | $ | (0.70 | ) | $ | (1.01 | ) | |||||||
| Diluted |
$ | 0.35 | $ | 0.44 | $ | 0.14 | $ | (0.70 | ) | $ | (1.01 | ) | |||||||
| Weighted average shares used in computing net income (loss) per share: |
|||||||||||||||||||
| Basic |
42,252 | 44,150 | 44,146 | 43,795 | 37,267 | ||||||||||||||
| Diluted |
43,857 | 45,676 | 45,475 | 43,795 | 37,267 | ||||||||||||||
| December 31, | |||||||||||||||||||
| 2008 | 2007 | 2006 | 2005 | 2004 | |||||||||||||||
| Balance sheet data: |
|||||||||||||||||||
| Cash and cash equivalents |
$ | 153,158 | $ | 86,567 | $ | 16,386 | $ | 95,651 | $ | 1,379 | |||||||||
| Marketable securities |
36,937 | 118,504 | 188,014 | 117,001 | 98,018 | ||||||||||||||
| Working capital |
170,522 | 184,717 | 170,460 | 181,817 | 91,860 | ||||||||||||||
| Total assets |
193,436 | 207,625 | 208,456 | 215,795 | 101,192 | ||||||||||||||
| Deferred program fee revenue |
82,502 | 96,849 | 120,087 | 146,288 | — | ||||||||||||||
| Total liabilities |
89,150 | 103,711 | 130,541 | 152,435 | 7,796 | ||||||||||||||
| Total stockholders’ equity |
104,286 | 103,914 | 77,915 | 63,360 | 93,396 | ||||||||||||||
| (1) | Includes stock option expense after adoption of the Financial Statement Accounting Board, Statement No. 123R on January 1, 2006. |
28
Table of Contents
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
This discussion and analysis should be read in conjunction with our financial statements and accompanying notes included elsewhere in this report. Operating results are not necessarily indicative of results that may occur in future periods.
Overview
We are a biopharmaceutical company that develops novel drugs. In June 2008, we filed an NDA for Remoxy with the FDA. In August 2008, we and King announced that an NDA for Remoxy was accepted by the FDA and granted Priority Review. A Priority Review generally results in a reduction in the time for FDA review of an NDA from 12 months to approximately 6 months from the date of the NDA submission. In December 2008, we received a Complete Response Letter for our NDA for Remoxy in which the FDA determined that the NDA was not approved. The FDA indicated additional non-clinical data will be required to support the approval of REMOXY. The FDA has not requested or recommended additional clinical efficacy studies prior to approval. We plan to meet with the FDA in the second quarter of 2009 regarding the NDA for Remoxy. We believe this FDA meeting will provide us with a more reliable context in which to make projections about Remoxy.
We also have the following investigational drug candidates in clinical programs:
| • | PTI-202 and PTI-721, which are proprietary, abuse-resistant forms of opioid drugs. |
| • | A novel radio-labeled monoclonal antibody drug candidate to treat metastatic melanoma, a deadly form of skin cancer. |
We and King are engaged in a strategic alliance to develop and commercialize Remoxy, PTI-202, PTI-721 and another abuse-resistant opioid painkiller. King is also obligated to fund development expenses incurred by us pursuant to the collaboration agreement.
All of our collaboration, contract and milestone revenues are recognized pursuant to our strategic alliance with King. King made an upfront cash payment of $150.0 million to us at the closing of this strategic alliance in 2005. King made milestone payments to us of $15.0 million related to acceptance by the FDA of the NDA for Remoxy and $5.0 million in 2008 in connection with the acceptance by the FDA of the investigational new drug application, or IND for PTI-721 and $5.0 million in 2006 in connection with the acceptance by the FDA of the IND for PTI-202.
We could also receive from King up to $125.0 million in additional milestone payments in the course of clinical development of Remoxy, PTI-202, PTI-721 and other abuse-resistant opioid painkillers under the strategic alliance. Subject to certain limitations, King is also obligated to fund certain development expenses incurred by us pursuant to the collaboration agreement. King is obligated to fund the commercialization expenses of, and has the exclusive right to market and sell, drugs developed in connection with the strategic alliance. King is obligated to pay us a 20% royalty on net sales of drugs developed in connection with the strategic alliance, except as to the first $1.0 billion in net sales of such drugs, for which the royalty is set at 15%.
Although we were profitable in 2006, 2007 and 2008 based on payments received from King and interest income, we have yet to generate any revenues from product sales. Through December 31, 2008, we have recorded an accumulated deficit of approximately $114.1 million. These losses have resulted principally from costs incurred in connection with research and development activities, salaries and other personnel-related costs and general corporate expenses. Research and development activities include costs of preclinical and clinical trials as well as clinical supplies associated with our drug candidates. Salaries and other personnel-related costs include non-cash stock-based compensation associated with options granted to employees and non-employees. Our operating results may fluctuate substantially from period to period as a result of the timing and enrollment rates of clinical trials for our drug candidates and our need for clinical supplies.
29
Table of Contents
We expect to continue to use significant cash resources in our operations for the next several years. Our cash requirements for operating activities and capital expenditures will increase substantially in the future as we:
| • | continue to conduct preclinical and clinical trials for our drug candidates; |
| • | seek regulatory approvals for our drug candidates; |
| • | develop, formulate, manufacture and commercialize our drug candidates; |
| • | implement additional internal systems and develop new infrastructure; |
| • | acquire or in-license additional products or technologies, or expand the use of our technology; |
| • | maintain, defend and expand the scope of our intellectual property; and |
| • | hire additional personnel. |
Product revenue will depend on our ability to receive regulatory approvals for, and successfully market, our drug candidates. If our development efforts result in regulatory approval and successful commercialization of our drug candidates, we will generate revenue from direct sales of our drugs and/or, if we license our drugs to future collaborators, from the receipt of license fees and royalties from sales of licensed products. We conduct our research and development programs through a combination of internal and collaborative programs. We rely on arrangements with universities, our collaborators, contract research organizations and clinical research sites for a significant portion of our product development efforts.
We focus substantially all our research and development efforts on the research and development of opioid drugs for the treatment of pain. The following table summarizes expenses by category for research and development efforts (in thousands):
| Years Ended December 31, | |||||||||
| 2008 | 2007 | 2006 | |||||||
| Compensation |
$ | 15,117 | $ | 11,126 | $ | 9,746 | |||
| Contractor fees (1) |
22,112 | 26,405 | 30,367 | ||||||
| Supplies (2) |
3,668 | 6,325 | 4,317 | ||||||
| Other common costs (3) |
4,920 | 3,874 | 2,373 | ||||||
| $ | 45,817 | $ | 47,730 | $ | 46,803 | ||||
| (1) | Contractor fees generally include expenses for preclinical studies and clinical trials. |
| (2) | Supplies generally include costs for formulation and manufacturing activities. |
| (3) | Other generally includes the allocation of common costs such as facilities. |
Our technology has been applied across certain of our portfolio of drug candidates. Data, know-how, personnel, clinical results, research results and other matters related to the research and development of any one of our drug candidates also relate to, and further the development of, our other drug candidates. For example, we expect that results of non-clinical studies, such as pharmacokinetics, toxicology and other studies, regarding certain components of our drug candidate Remoxy to be applicable to the other potential drug candidates that may arise out of our collaboration with King since all such potential drug candidates are expected to utilize such components. As a result, costs allocated to a specific drug candidate may not necessarily reflect the actual costs surrounding research and development of that drug candidate due to cross application of the foregoing.
We are developing a novel antibody technology that may transform how metastatic melanoma is treated. We spent approximately $2.8, $3.2 million and $3.0 million on this technology, primarily in contractor fees and supplies, during 2008, 2007 and 2006, respectively. In 2007, we announced that we licensed certain technology and are developing this technology for hemophilia and pain management. We spent approximately $6.0 and $1.0 million, primarily in contractor fees and supplies during 2008 and 2007 on this technology.
30
Table of Contents
Estimating the dates of completion of clinical development, and the costs to complete development, of our drug candidates would be highly speculative, subjective and potentially misleading. Pharmaceutical products take a significant amount of time to research, develop and commercialize. The clinical trial portion of the development of a new drug alone usually spans several years. We expect to reassess our future research and development plans based on our review of data we receive from our current research and development activities. The cost and pace of our future research and development activities are linked and subject to change.
Critical Accounting Policies
The preparation of our financial statements in accordance with United States generally accepted accounting principles requires us to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenues, expenses and interest income in our financial statements and accompanying notes. We evaluate our estimates on an ongoing basis, including those estimates related to contract agreements, research collaborations and investments. We base our estimates on historical experience and various other assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions. The following items in our financial statements require significant estimates and judgments:
| • | Expenses for clinical trials. Expenses for clinical trials are incurred from planning through patient enrollment to reporting of the underlying data. We estimate expenses incurred for clinical trials that are in process based on patient enrollment and based on clinical data collection and management. Costs that are associated with patient enrollment are recognized as each patient in the clinical trial completes enrollment. Estimated clinical trial costs related to enrollment can vary based on numerous factors, including expected number of patients in trials, the number of patients that do not complete participation in a trial, and when a patient drops out of a trial. Information about patient enrollment can become available significantly after we report our expenses for clinical trials, in which case we would change our estimate of the remaining cost of a trial. Costs that are based on clinical data collection and management are recognized based on estimates of unbilled goods and services received. In the event of early termination of a clinical trial, we would accrue an amount based on estimates of the remaining non-cancelable obligations associated with winding down the clinical trial. |
| • | Stock-based compensation. The Financial Accounting Standards Board, or FASB, Statement No. 123 (revised 2004), Share-Based Payment, or SFAS 123R, requires companies to recognize expense in the income statement for the fair value of all share-based payments to employees and directors, including grants of employee stock options and other share based awards. We adopted SFAS 123R on January 1, 2006 using the modified-prospective transition method. We record compensation expense for all awards granted after the date of adoption and for the unvested portion of previously granted awards that remain outstanding at the date of adoption. We use the Black-Scholes option valuation model, or Black-Scholes and use the single-option award approach and straight-line attribution method for stock options granted since January 1, 2006. Using this approach, the compensation cost is amortized on a straight-line basis over the vesting period of each respective stock option, generally four years. |
We estimate forfeitures when recognizing expense under SFAS 123R and adjust this estimate periodically based on the extent to which future actual forfeitures differ, or are expected to differ, from such estimates. Accordingly, we have estimated forfeiture percentages for the unvested portion of previously granted awards that remain outstanding at the date of adoption and for awards granted subsequent to the date of adoption.
We have granted share-based awards that vest upon achievement of certain performance criteria, or Performance Awards. The value of these awards is the product of the number of shares of our common stock to be issued under the award multiplied by the fair market value of a share of our common stock on the date of grant. These awards include future performance conditions. We estimate an implicit service period for achieving these performance conditions. Performance Awards vest and common stock
31
Table of Contents
is issued on achieving performance conditions. We recognize stock-based compensation expense for Performance Awards when we conclude that achieving a performance condition is probable, consistent with FASB Statement No. 5, Accounting for Contingencies. We periodically review and update as appropriate our estimates of the implicit service periods and the likelihood of achieving the performance conditions.
| • | Revenue recognition and deferred program fee revenue. In connection with our strategic alliance with King we recognize program fee revenue, collaboration revenue and milestone revenue. Program fee revenue is derived from the upfront payment from King received in December 2005 and is recognized ratably over our estimate of the development period of four drug candidates expected to be developed under the strategic alliance with King. Of those drug candidates, Remoxy has completed a Phase III clinical trial, one drug candidate is in Phase I clinical trials and two potential drug candidates are at the pre-clinical stage. We currently estimate the development period for all four expected drug candidates to extend through September 2014. We review the estimated development period on a quarterly basis and change it if appropriate based upon our latest expectations. For example, in the fourth quarter of 2007 we determined that the development period should be extended from the third quarter of 2011 based upon recent clinical developments and updated communications with King regarding product development plans. Collaboration revenues from reimbursement of development expenses are generally recognized when King has completed its review of the expenses invoiced to them. King is obligated to pay us milestone payments contingent upon the achievement of certain substantive events in the clinical development of Remoxy and the other abuse-resistant opioid painkillers under the strategic alliance. We recognize milestone payments from King as revenue when we achieve the underlying developmental milestone as the milestone payments are not dependent upon any other future activities or achievement of any other future milestones and the achievement of each of the developmental milestones were substantively at risk and contingent at the effective date of the collaboration. Substantial effort is involved in achieving each of the developmental milestones. These milestones represent the culmination of discrete earnings processes and the amount of each milestone payment is reasonable in relation with the level of effort associated with the achievement of the milestone. Each milestone payment is non-refundable and non-creditable when made. The ongoing research and development services being provided to King under the collaboration are priced at fair value based upon the reimbursement of expenses incurred pursuant to the collaboration with King. |
| • | Taxes. We make estimates and judgments in determining our provision for (benefit from) income taxes. We have accumulated significant deferred tax assets. Deferred income taxes reflect the tax effects of net operating loss and tax credit carryovers and temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Realization of deferred tax assets is dependent upon future earnings, if any. We are uncertain of the timing and amount of any future earnings. Accordingly, except for $1.9 million of net deferred tax assets recognized on our balance sheet included in other assets as of December 31, 2008, we offset the net deferred tax assets with a valuation allowance. We may in the future determine that more of our deferred tax assets will likely be realized, in which case we will reduce our valuation allowance in the quarter in which such determination is made. If the valuation allowance is reduced, we may recognize a benefit from income taxes in our statement of operations in that period. |
Results of Operations
Years Ended December 31, 2008 and 2007
Revenue—Program fee revenue
King paid us a $150.0 million upfront fee in connection with the closing of our strategic alliance with King in December 2005. Revenues recognized from amortization of this upfront fee were 14.3 million and $23.2 million in the years ended December 31, 2008 and 2007, respectively. The decrease of $8.9 million from 2007 to 2008 is due to the extension of the development period over which we recognize the program fee revenue in the
32
Table of Contents
fourth quarter of 2007 based upon recent clinical developments and communications with King regarding product development plans. We expect to recognize the rest of the program fee ratably over our estimate of the remainder of the development period under the strategic alliance with King. We currently estimate the development period for all four expected drug candidates to extend through September 2014.
Revenue—Collaboration revenue
Collaboration revenues were $29.4 million and $42.7 million in the years ended December 31, 2008 and 2007, respectively. These revenues related to reimbursement of our development expenses incurred pursuant to the King strategic alliance. The decrease of $13.3 million in 2008 was primarily because 2007 revenue included recognition of approximately $10.4 million in costs incurred by us between September and December 2006 that were subject to completion of King’s review. King reviewed and paid for these expenses in 2007. We incurred expenses of $3.8 million in 2007, which were reimbursement by King in 2008. We incurred expenses of $1.1 million in 2008 for which we expect King to complete their review and to reimburse us in 2009.
We expect the amount and timing of collaboration revenue to fluctuate in relation to the amount and timing of the underlying research and development expenses, as well as the timing of completion of King’s review of submitted expenses.
Revenue—Milestone revenue
Milestone revenue was $20.0 million in the year ended December 31, 2008. King made a milestone payments of $15.0 million to us on acceptance of the NDA for Remoxy by the FDA and $5.0 million in connection with the acceptance by the FDA of the IND for PTI-202.
Research and Development Expense
Research and development expense consists primarily of costs of drug development work associated with our drug candidates, including:
| • | preclinical testing, |
| • | clinical trials, |
| • | clinical supplies and related formulation and design costs, and |
| • | salaries and other personnel-related expenses. |
Research and development expense decreased to $45.8 million from $47.7 million in the years ended December 31, 2008 and 2007, respectively. The decrease was primarily due to decreases in clinical and development activities for Remoxy partially offset by increased activities in metastatic melanoma, hemophilia and other projects. Research and development expenses included non-cash stock related compensation costs of $6.1 million and $3.7 million in the twelve months ended December 31, 2008 and 2007, respectively. Non-cash stock related compensation costs in 2008 included $1.7 million associated with vesting of Performance Awards.
We expect research and development expenses to fluctuate over the next several years as we continue our development efforts. We expect our development efforts to result in our drug candidates progressing through various stages of clinical trials, including current and potential clinical trials for our other abuse-resistant drug candidates, as well as further clinical development of our product candidates in metastatic melanoma and hemophilia. King is obligated to reimburse development expenses for our abuse-resistant drug candidates pursuant to our collaboration. Our research and development expenses may fluctuate from period to period due to the timing and scope of our development activities and the results of clinical trials and preclinical studies.
33
Table of Contents
General and Administrative Expense
General and administrative expenses consist primarily of compensation and other general corporate expenses. General and administrative expenses increased to $9.2 million from $8.1 million in the years ended December 31, 2008 and 2007, respectively. The increase was primarily due to increases in non-cash stock-related compensation costs. General and administrative expenses included non-cash stock related compensation costs of $4.1 million and $2.6 million in the twelve months ended December 31, 2008 and 2007, respectively. Non-cash stock related compensation costs in 2008 included $1.6 million associated with vesting of Performance Awards. We expect general and administrative expenses to increase over the next several years in connection with support of precommercialization and commercialization activities for our drug candidates. The increase may fluctuate from period to period due to the timing and scope of these activities and the results of clinical trials and preclinical studies.
Interest and Other Income, Net
Interest and other income, net, decreased to $6.0 million from $10.1 million for the years ended December 31, 2008 and 2007, respectively, primarily due to decreases in prevailing interest rates on investments in marketable securities and, to a lesser extent, decreased average balances of marketable securities. We expect our interest income to decrease in the future as we use cash to fund our operations.
Benefit from Income Taxes
In 2008 we had an income tax benefit of $0.6 million comprised of a federal tax benefit of $0.8 million, net of a provision for state taxes of $0.2 million. The federal income tax benefit is primarily due to the recognition in 2008 of previously generated net operating losses and tax credits. We did not provide for income taxes in 2007 because we did not have taxable income in 2007, primarily due to all of our program fee revenue having been recognized for tax purposes in prior years.
Years Ended December 31, 2007 and 2006
Revenue—Program fee revenue
King paid us a $150.0 million upfront fee in connection with the closing of our strategic alliance with King in December 2005. Revenues recognized from amortization of this upfront fee were $23.2 million and $26.2 million in the years ended December 31, 2007 and 2006, respectively. The decrease of $3.0 million in 2007 is due to the extension of the development period over which we recognize the program fee revenue in the fourth quarter of 2007 based upon recent clinical developments and communications with King regarding product development plans.
Revenue—Collaboration revenue
Collaboration revenues were $42.7 million and $22.7 million in the years ended December 31, 2007 and 2006, respectively. These revenues related to reimbursement of our development expenses incurred pursuant to the King strategic alliance. The increase of $20.0 million in 2007 was primarily because in 2006 we deferred recognition of approximately $10.4 million in costs incurred by us between September and December 2006 that were subject to completion of King’s review. King reviewed and paid for these expenses in 2007. We incurred expenses of $3.8 million in 2007 for which King completed their review and reimbursed us in 2008.
Revenue—Milestone revenue
We did not have any milestone revenue in the year ended December 31, 2007. Milestone revenue was $5.0 million for the year end December 31, 2006. In connection with the acceptance by the FDA of the investigational new drug application for PTI-202, King made a milestone payment to us of $5.0 million.
34
Table of Contents
Research and Development Expense
Research and development expense consists primarily of costs of drug development work associated with our drug candidates, including:
| • | preclinical testing, |
| • | clinical trials, |
| • | clinical supplies and related formulation and design costs, and |
| • | salaries and other personnel-related expenses. |
Research and development expense increased to $47.7 million from $46.8 million in the years ended December 31, 2007 and 2006, respectively. The increase was primarily due to increases in clinical and development activities for Remoxy, metastatic melanoma and hemophilia. Research and development expenses included non-cash stock related compensation costs of $3.7 million and $3.8 million in the twelve months ended December 31, 2007 and 2006, respectively.
General and Administrative Expense
General and administrative expenses consist primarily of compensation and other general corporate expenses. General and administrative expenses increased to $8.1 million from $7.7 million in the years ended December 31, 2007 and 2006, respectively. The increase was primarily due to increases in non-cash stock-related compensation costs associated with the adoption of SFAS 123R. General and administrative expenses included non-cash stock related compensation costs of $2.6 million and $2.7 million in the twelve months ended December 31, 2007 and 2006, respectively.
Interest and Other Income, Net
Interest and other income net, increased to $10.1 million from $9.7 million for the years ended December 31, 2007 and 2006, respectively, primarily due to increases in average balances of marketable securities and, to a lesser extent, increases in prevailing interest rates on investments in marketable securities.
Provision for Income Taxes
We did not provide for income taxes in 2007 because we did not have taxable income in 2007, primarily due to all of our program fee revenue having been recognized for tax purposes in prior years.
In 2005, King made an upfront cash payment of $150.0 million to us in connection with our strategic alliance. We had taxable income for 2006 primarily due to the recognition in 2006 of $146.3 million of the upfront cash payment for income tax purposes. We reduced our taxable income for 2006 with deductions related to a combination of our net operating losses and tax credits from prior years.
Liquidity and Capital Resources
Since inception, we have financed our operations primarily through public and private stock offerings, payments received under our strategic alliance with King, and interest earned on our investments. We intend to continue to use our capital resources to fund research and development activities, capital expenditures, working capital requirements and other general corporate purposes. As of December 31, 2008, cash, cash equivalents and marketable securities were $190.1 million.
Net cash provided by operating activities was $11.2 million for the year ended December 31, 2008 compared to net cash provided by operating activities of $2.2 million for the year ended December 31, 2007. The change was primarily due to the increase in 2008 of $20.0 million received in milestone payments offset in part by a $13.3 million decrease in collaboration revenue.
35
Table of Contents
Net cash provided by investing activities was $80.2 million in the year ended December 31, 2008 and $69.1 million for the year ended December 31, 2007. Investing activities for both years consisted primarily of the purchase and sale of marketable securities. Our investing activities to purchase property, equipment and leasehold improvements used no cash for the year ended December 31, 2008 and $0.7 million for the year ended December 31, 2007. In the year ended December 31, 2008, we wrote-off our property, equipment and leasehold improvements and accumulated depreciation and amortization related to our lease of office space in South San Francisco because we no longer use that office space. We expect to continue to invest in our infrastructure to support our operations.
Net cash used in financing activities was $24.8 million in the year ended December 31, 2008 and $1.1 million in the year ended December 31, 2007. In 2008, we completed a stock buyback plan for the repurchase of up to $30.0 million of our common stock. We used cash of $26.2 million in 2008 and $3.8 million in 2007 to purchase our common stock on the open market. Other cash from financing activities in both 2008 and 2007 consisted primarily of proceeds received from the issuance of our common stock pursuant to stock option exercises and stock purchased by employees pursuant to our 2000 Employee Stock Purchase Plan.
We have $43.5 million of total deferred tax assets at December 31, 2008. Realization of these deferred tax assets is dependent on future earnings, if any. We are uncertain about the timing and amount of any future earnings. We have concluded that it is more likely than not that such deferred tax assets will not be realized. Accordingly, except for $1.9 million of deferred tax assets included in other assets as of December 31, 2008, we offset the deferred tax asset with a valuation allowance. There is a high degree of uncertainty regarding the timing of future cash outflows associated with our FASB Interpretation No. 48, “Accounting for Uncertainty in Income Taxes,” or Interpretation 48 liabilities. Our net Interpretation 48 liability at December 31, 2008 does not result in a material contractual obligation.
We currently lease approximately 41,200 square feet of general office space pursuant to non-cancelable operating leases that will expire in 2010 and 2012. Future minimum lease payments for our leases are as follows for the years ended December 31, (in thousands):
| 2009 | 2010 | 2011 | 2012 | Total | |||||||||||
| Future minimum lease payments |
$ | 743 | $ | 713 | $ | 570 | $ | 339 | $ | 2,365 | |||||
We believe that our facilities are adequate and suitable for our current needs.
We have license agreements that require us to make milestone payments upon the successful achievement of milestones, including clinical milestones. Our license agreements also require us to pay certain royalties to our licensors if we succeed in fully commercializing products under these license agreements. All of these potential future payments are cancelable as of December 31, 2008. Our formulation agreement with Durect Corporation obligates us to make certain milestone payments upon achieving clinical milestones and regulatory milestones. King is obligated to reimburse us for any of our milestone payments and royalty payments to Durect Corporation.
We have an accumulated deficit of $114.1 million at December 31, 2008. We expect our cash requirements to be significant in the future. The amount and timing of our future cash requirements will depend on regulatory and market acceptance of our drug candidates and the resources we devote to researching and developing, formulating, manufacturing, commercializing and supporting our products. We believe that our current resources should be sufficient to fund our operations for at least the next 12 months. We may seek additional future funding through public or private financing within this timeframe, if such funding is available and on terms acceptable to us.
Off-balance Sheet Arrangements
As of December 31, 2008, we did not have any relationships with unconsolidated entities or financial partnerships, such as entities often referred to as structured finance or special purpose entities, which would have
36
Table of Contents
been established for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or limited purposes. In addition, we do not engage in trading activities involving non-exchange traded contracts. Therefore, we are not materially exposed to financing, liquidity, market or credit risk that could arise if we had engaged in these relationships. We do not have relationships or transactions with persons or entities that derive benefits from their non-independent relationship with us or our related parties.
| Item 7A. | Quantitative and Qualitative Disclosures about Market Risk |
The primary objective of our cash investment activities is to preserve principal while at the same time maximizing the income we receive from our investments without significantly increasing risk. Some of the securities that we invest in may be subject to market risk. This means that a change in prevailing interest rates may cause the principal amount of the investment to fluctuate. For example, if we hold a security that was issued with a fixed interest rate at the then-prevailing rate and the interest rate later rises, the principal amount of our investment will probably decline. A hypothetical 50 basis point increase in interest rates reduces the fair value of our available-for-sale securities at December 31, 2008 by approximately $0.1 million. To minimize this risk in the future, we intend to maintain our portfolio of cash equivalents and marketable securities in a variety of securities, including commercial paper, government and non-government debt securities and/or money market funds that invest in such securities. We have no holdings of derivative financial or commodity instruments. As of December 31, 2008, our investments consisted of investments in corporate and government notes and obligations or in money market accounts and checking funds with variable market rates of interest. We believe our credit risk is immaterial.
37
Table of Contents
| Item 8. | Financial Statements and Supplementary Data |
| Page | ||
| 39 | ||
| 40 | ||
| 41 | ||
| 42 | ||
| 43 | ||
| 44 | ||
38
Table of Contents
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders
Pain Therapeutics, Inc.
We have audited the accompanying balance sheets of Pain Therapeutics, Inc. as of December 31, 2008 and 2007, and the related statements of operations, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2008. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Pain Therapeutics, Inc. at December 31, 2008 and 2007, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2008, in conformity with U.S. generally accepted accounting principles.
As discussed in Notes 8 and 9 to the financial statements, in 2007, Pain Therapeutics, Inc. changed its methods of accounting for uncertainty in income taxes and sabbatical leave in accordance with guidance provided in Statement of Financial Accounting Standards No. 48, “Accounting for Uncertainty in Income Taxes an interpretation of FASB Statement No. 109” and Emerging Issues Task Force Issue No. 06-2, “Accounting for Sabbatical Leave and Other Similar Benefits Pursuant to FASB Statement No. 43.”
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Pain Therapeutics, Inc.’s internal control over financial reporting as of December 31, 2008, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated February 11, 2009 expressed an unqualified opinion thereon.
ERNST & YOUNG LLP
San Jose, California
February 11, 2009
39
Table of Contents
BALANCE SHEETS
(in thousands except share and per share data)
| December 31, | ||||||||
| 2008 | 2007 | |||||||
| ASSETS | ||||||||
| Current assets |
||||||||
| Cash and cash equivalents |
$ | 153,158 | $ | 86,567 | ||||
| Marketable securities |
36,937 | 118,504 | ||||||
| Other current assets |
541 | 303 | ||||||
| Total current assets |
190,636 | 205,374 | ||||||
| Property and equipment, net |
774 | 1,607 | ||||||
| Other assets |
2,026 | 644 | ||||||
| Total assets |
$ | 193,436 | $ | 207,625 | ||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| Current liabilities |
||||||||
| Accounts payable |
$ | 2,216 | $ | 3,624 | ||||
| Accrued development expense |
1,029 | 817 | ||||||
| Deferred program fee revenue—current portion |
14,348 | 14,348 | ||||||
| Other accrued liabilities |
2,521 | 1,868 | ||||||
| Total current liabilities |
20,114 | 20,657 | ||||||
| Non-current liabilities |
||||||||
| Deferred program fee revenue—non-current portion |
68,154 | 82,501 | ||||||
| Deferred tax liabilities |
882 | 553 | ||||||
| Total liabilities |
89,150 | 103,711 | ||||||
| Commitments and contingencies |
||||||||
| Stockholders’ equity |
||||||||
| Preferred stock, $.001 par value; 10,000,000 shares authorized, none issued and outstanding |
— | — | ||||||
| Common stock, $.001 par value; 120,000,000 shares authorized; 42,068,752 and 44,305,103 shares issued and outstanding in 2008 and 2007, respectively |
42 | 44 | ||||||
| Additional paid-in-capital |
218,021 | 221,415 | ||||||
| Accumulated other comprehensive income |
325 | 584 | ||||||
| Accumulated deficit |
(114,102 | ) | (118,129 | ) | ||||
| Total stockholders’ equity |
104,286 | 103,914 | ||||||
| Total liabilities and stockholders’ equity |
$ | 193,436 | $ | 207,625 | ||||
See accompanying notes to financial statements.
40
Table of Contents
STATEMENTS OF OPERATIONS
(in thousands except per share data)
| Years ended December 31, | |||||||||||
| 2008 | 2007 | 2006 | |||||||||
| Revenue |
|||||||||||
| Collaboration revenue |
$ | 29,377 | $ | 42,746 | $ | 22,717 | |||||
| Program fee revenue |
14,348 | 23,238 | 26,201 | ||||||||
| Milestone revenue |
20,000 | — | 5,000 | ||||||||
| Total revenue |
63,725 | 65,984 | 53,918 | ||||||||
| Operating expenses |
|||||||||||
| Research and development |
45,817 | 47,730 | 46,803 | ||||||||
| General and administrative |
9,196 | 8,085 | 7,668 | ||||||||
| Total operating expenses |
55,013 | 55,815 | 54,471 | ||||||||
| Operating income (loss) |
8,712 | 10,169 | (553 | ) | |||||||
| Interest and other income, net |
6,018 | 10,136 | 9,668 | ||||||||
| Income before provision for (benefit from) income taxes |
14,730 | 20,305 | 9,115 | ||||||||
| Provision for (benefit from) income taxes |
(617 | ) | — | 2,927 | |||||||
| Net income |
$ | 15,347 | $ | 20,305 | $ | 6,188 | |||||
| Net income per share |
|||||||||||
| Basic |
$ | 0.36 | $ | 0.46 | $ | 0.14 | |||||
| Diluted |
$ | 0.35 | $ | 0.44 | $ | 0.14 | |||||
| Weighted-average shares used in computing net income per share |
|||||||||||
| Basic |
42,252 | 44,150 | 44,146 | ||||||||
| Diluted |
43,857 | 45,676 | 45,475 | ||||||||
See accompanying notes to financial statements.
41
Table of Contents
STATEMENT OF STOCKHOLDERS’ EQUITY
(in thousands except share data)
| Common stock | Additional paid-in capital |
Accumulated other comprehensive income (loss) |
Accumulated deficit |
Total stockholders’ equity |
|||||||||||||||||||
| Shares | Par value | ||||||||||||||||||||||
| Balance at December 31, 2005 |
43,936,088 | $ | 44 | $ | 206,489 | $ | (479 | ) | $ | (142,694 | ) | $ | 63,360 | ||||||||||
| Issuance of common stock pursuant to exercise of stock options |
304,180 | — | 1,312 | — | — | 1,312 | |||||||||||||||||
| Issuance of common stock related to employee stock purchase plan |
73,735 | — | 330 | — | — | 330 | |||||||||||||||||
| Compensation with respect to non-employee option grants |
— | — | 242 | — | — | 242 | |||||||||||||||||
| Compensation with respect to employee option grants |
— | — | 6,228 | — | — | 6,228 | |||||||||||||||||
| Tax benefits from the exercise of options |
— | — | 148 | — | — | 148 | |||||||||||||||||
| Net unrealized gains on investment in marketable securities |
— | — | — | 107 | — | 107 | |||||||||||||||||
| Net income |
— | — | — | — | 6,188 | 6,188 | |||||||||||||||||
| Comprehensive income |
6,295 | ||||||||||||||||||||||
| Balance at December 31, 2006 |
44,314,003 | 44 | 214,749 | (372 | ) | (136,506 | ) | 77,915 | |||||||||||||||
| Issuance of common stock pursuant to exercise of stock options |
400,442 | — | 2,284 | — | — | 2,284 | |||||||||||||||||
| Issuance of common stock related to employee stock purchase plan |
71,358 | — | 368 | — | — | 368 | |||||||||||||||||
| Compensation with respect to non-employee option grants |
— | — | 437 | — | — | 437 | |||||||||||||||||
| Compensation with respect to employee option grants |
— | — | 5,857 | — | — | 5,857 | |||||||||||||||||
| Accrued sabbatical benefits |
— | — | — | — | (415 | ) | (415 | ) | |||||||||||||||
| Purchase of stock pursuant to the stock repurchase plan |
(480,700 | ) | — | (2,280 | ) | — | (1,513 | ) | (3,793 | ) | |||||||||||||
| Net unrealized gains on investment in marketable securities |
— | — | — | 956 | — | 956 | |||||||||||||||||
| Net income |
— | — | — | — | 20,305 | 20,305 | |||||||||||||||||
| Comprehensive income |
21,261 | ||||||||||||||||||||||
| Balance at December 31, 2007 |
44,305,103 | 44 | 221,415 | 584 | (118,129 | ) | 103,914 | ||||||||||||||||
| Issuance of common stock pursuant to exercise of stock options |
819,058 | 1 | 638 | — | — | 639 | |||||||||||||||||
| Issuance of common stock related to employee stock purchase plan |
73,354 | — | 462 | — | — | 462 | |||||||||||||||||
| Compensation with respect to non-employee option grants |
— | — | 128 | — | — | 128 | |||||||||||||||||
| Compensation with respect to employee option grants and share based awards |
— | — | 10,077 | — | — | 10,077 | |||||||||||||||||
| Purchase of stock pursuant to the stock repurchase plan |
(3,128,763 | ) | (3 | ) | (14,884 | ) | — | (11,320 | ) | (26,207 | ) | ||||||||||||
| Tax benefits from the exercise of options |
— | — | 185 | — | — | 185 | |||||||||||||||||
| Net unrealized losses on investments in marketable securities |
— | — | — | (259 | ) | — | (259 | ) | |||||||||||||||
| Net income |
— | — | — | — | 15,347 | 15,347 | |||||||||||||||||
| Comprehensive income |
15,088 | ||||||||||||||||||||||
| Balance at December 31, 2008 |
42,068,752 | $ | 42 | $ | 218,021 | $ | 325 | $ | (114,102 | ) | $ | 104,286 | |||||||||||
See accompanying notes to financial statements.
42
Table of Contents
STATEMENTS OF CASH FLOWS
(in thousands)
| Years ended December 31, | ||||||||||||
| 2008 | 2007 | 2006 | ||||||||||
| Cash flows provided by (used in) operating activities: |
||||||||||||
| Net income |
$ | 15,347 | $ | 20,305 | $ | 6,188 | ||||||
| Adjustments to reconcile net income to net cash provided by (used in) operating activities: |
||||||||||||
| Non-cash stock based compensation |
10,205 | 6,294 | 6,470 | |||||||||
| Depreciation and amortization |
452 | 356 | 345 | |||||||||
| Non-cash net interest income |
1,073 | 679 | (1,858 | ) | ||||||||
| Loss on disposal of property and equipment |
381 | — | 38 | |||||||||
| Changes in operating assets and liabilities: |
||||||||||||
| Deferred program fee revenue |
(14,348 | ) | (23,238 | ) | (26,201 | ) | ||||||
| Other current assets |
(238 | ) | 2,411 | (1,202 | ) | |||||||
| Other assets |
(1,060 | ) | (86 | ) | — | |||||||
| Accounts payable |
(1,408 | ) | 2,639 | (13 | ) | |||||||
| Accrued development expense |
212 | (4,960 | ) | 1,316 | ||||||||
| Income taxes payable |
— | (2,779 | ) | 2,779 | ||||||||
| Tax benefits from equity-based compensation plans |
185 | — | 148 | |||||||||
| Excess tax benefits from equity-based compensation plans |
(298 | ) | — | (62 | ) | |||||||
| Other accrued liabilities |
660 | 610 | 225 | |||||||||
| Net cash provided by (used in) operating activities |
11,163 | 2,231 | (11,827 | ) | ||||||||
| Cash flows provided by (used in) investing activities: |
||||||||||||
| Purchase of property and equipment |
— | (696 | ) | (94 | ) | |||||||
| Purchase of marketable securities |
(2,122 | ) | (138,394 | ) | (136,119 | ) | ||||||
| Sales of marketable securities |
52,887 | 199,681 | 27,261 | |||||||||
| Maturities of marketable securities |
29,471 | 8,500 | 39,810 | |||||||||
| Net cash provided by (used in) investing activities |
80,236 | 69,091 | (69,142 | ) | ||||||||
| Cash flows provided by (used in) financing activities: |
||||||||||||
| Excess tax benefits from equity-based compensation plans |
298 | — | 62 | |||||||||
| Proceeds from issuance of common stock, net |
1,101 | 2,652 | 1,642 | |||||||||
| Purchase of stock pursuant to the stock repurchase plan |
(26,207 | ) | (3,793 | ) | — | |||||||
| Net cash provided by (used in) financing activities |
(24,808 | ) | (1,141 | ) | 1,704 | |||||||
| Net increase (decrease) in cash and cash equivalents |
66,591 | 70,181 | (79,265 | ) | ||||||||
| Cash and cash equivalents at beginning of the year |
86,567 | 16,386 | 95,651 | |||||||||
| Cash and cash equivalents at end of the year |
$ | 153,158 | $ | 86,567 | $ | 16,386 | ||||||
| Supplemental cash flow information: |
||||||||||||
| Cash paid for income taxes |
$ | — | $ | 2,800 | $ | — | ||||||
See accompanying notes to financial statements.
43
Table of Contents
NOTES TO FINANCIAL STATEMENTS
1. Business
Pain Therapeutics, Inc. is a biopharmaceutical company that develops novel drugs. We have four drug candidates in clinical programs, including Remoxy, PTI-202, PTI-721 and a novel radio-labeled monoclonal antibody to treat metastatic melanoma. We are also working on a new treatment for patients with hemophilia.
Although we were profitable in 2006, 2007 and 2008 based on payments from King Pharmaceuticals, Inc., or King, and interest income, in the course of our development activities, we have sustained cumulative operating losses. There are no assurances that additional financing will be available on favorable terms, or at all.
2. Summary of Significant Accounting Policies
Use of Estimates
The preparation of financial statements in conformity with generally accepted accounting principles requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Actual results could differ from those estimates.
Revenue Recognition and Deferred Program Fee Revenue
Revenue is recognized when there is persuasive evidence that an arrangement exists, delivery has occurred, the price is fixed and determinable, and collection is reasonably assured.
In November 2005, we and King announced a strategic alliance to develop and commercialize Remoxy and up to three other abuse-resistant opioid painkillers. In connection with the strategic alliance, we recognize program fee revenue, collaboration revenue and milestone revenue. Program fee revenue is derived from the upfront payment from King and is recognized ratably over our estimate of the development period of four drug candidates expected to be developed under the strategic alliance with King. We currently estimate the development period for all four expected drug candidates to extend through September 2014. We review the estimated development period on a quarterly basis and change it if appropriate based upon our latest expectations. In the fourth quarter of 2007 we determined that the development period should be extended from the third quarter of 2011 based upon recent clinical developments and communications to us from King regarding King’s development plans and forecast expenditures. Deferred program fee revenue represents the amount of the upfront payment that has not been recognized as revenue to date.
Collaboration revenues from reimbursement of development expenses are generally recognized when King has completed its review of the expenses invoiced to them.
King is obligated to pay us milestone payments contingent upon the achievement of certain substantive events in the clinical development of Remoxy and the other abuse-resistant opioid painkillers under the strategic alliance. We recognize milestone payments from King as revenue when we achieve the underlying developmental milestone as the milestone payments are not dependent upon any other future activities or achievement of any other future milestones and the achievement of each of the developmental milestones were substantively at risk and contingent at the effective date of the collaboration. Substantial effort is involved in achieving each of the developmental milestones. These milestones represent the culmination of discrete earnings processes and the amount of each milestone payment is reasonable in relation with the level of effort associated with the achievement of the milestone. Each milestone payment is non-refundable and non-creditable when made. The ongoing research and development services being provided to King under the collaboration are priced at fair value based upon the reimbursement of expenses incurred pursuant to the collaboration with King.
44
Table of Contents
PAIN THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
Cash, Cash Equivalents and Concentration of Credit Risk
We consider all highly liquid financial instruments with original maturities of three months or less to be cash equivalents. Cash and cash equivalents consist of cash maintained at two financial institutions and in money market funds. We believe the financial risks associated with these instruments are minimal. We have not experienced material losses from our investments in these securities.
Marketable Securities and Fair Value Measurements
Our marketable securities are held as “available-for-sale” pursuant to Statement of Financial Accounting Standards No. 115, “Accounting for Certain Investments in Debt and Equity Securities.” We classify these investments as current assets and carry them at fair value. Unrealized gains and losses are recorded as a separate component of stockholders’ equity as accumulated other comprehensive income. We recognize all realized gains and losses on our available-for-sale securities in interest and other income in the accompanying statement of operations. Our marketable securities are maintained at one financial institution and are governed by our investment policy as approved by our Board of Directors.
To date we have not recorded any impairment charges on marketable securities related to other-than-temporary declines in market value. We would recognize an impairment charge when the decline in the estimated fair value of a marketable security below the amortized cost is determined to be other-than-temporary. We consider various factors in determining whether to recognize an impairment charge, including the duration of time and the severity to which the fair value has been less than our amortized cost, any adverse changes in the investees’ financial condition and our intent and ability to hold the marketable security for a period of time sufficient to allow for any anticipated recovery in market value.
We adopted Statement of Financial Accounting Standards No. 157, “Fair Value Measurements”, or SFAS 157, on January 1, 2008. SFAS 157 defines fair value, establishes a framework and gives guidance regarding the methods used for measuring fair value and expands disclosure about such fair value measurements. The adoption of SFAS 157 did not have an impact on our results of operations or financial position.
We measure our cash equivalents and marketable securities at fair value on a recurring basis and have significant observable inputs where there are identical or comparable assets in the market to use in establishing our fair value measurements. We use significant observable inputs that include but are not limited to benchmark yields, reported trades, broker/dealer quotes and issuer spreads. Generally, the types of instruments we invest in are not traded on a market such as the NASDAQ Global Market. We consider these inputs to be Level 2 as defined by SFAS 157. We do not have any investments that would require inputs considered to be Level 3 as defined by SFAS 157. We use the bid price to establish fair value.
We adopted Statement of Financial Accounting Standards No. 159, “The Fair Value Option for Financial Assets and Financial Liabilities-including an amendment of FASB Statement No. 115”, or SFAS 159, on January 1, 2008. Under this statement, an entity may elect to use fair value to measure eligible items. We adopted SFAS 159 by electing not to apply the fair value election of SFAS 159. The adoption of this statement did not have an impact on our results of operations or financial condition.
Property and Equipment
Property and equipment are stated at cost, net of accumulated depreciation and amortization. Depreciation is calculated using the straight-line method over the estimated useful lives of the respective assets (generally two to five years). Leasehold improvements are amortized over the shorter of the estimated useful life of the assets or the lease term.
45
Table of Contents
PAIN THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
Impairment of Long-Lived Assets
We regularly perform reviews to determine if the carrying value of our long-lived assets is impaired. We consider facts or circumstances, either internal, or external that indicate that we may not recover the carrying value of the asset. No events or changes in circumstances have occurred with respect to our long-lived assets that would indicate that an impairment analysis should have been performed.
Business Segments
Statement of Financial Accounting Standards No. 131, Disclosures about Segments of an Enterprise and Related Information, requires an enterprise to report segment information based on how management internally evaluates the operating performance of its business units (segments). Our operations are confined to one business segment: the development of novel drugs.
Expenses for Clinical Trials
Research and development expense includes the cost of clinical trials. Expenses for clinical trials are incurred from planning through patient enrollment to reporting of the underlying data. We estimate expenses incurred for clinical trials that are in process based on patient enrollment and based on clinical data collection and management. Costs that are associated with patient enrollment are recognized as each patient in the clinical trial completes enrollment. Estimated clinical trial costs related to enrollment can vary based on numerous factors, including expected number of patients in trials, the number of patients that do not complete participation in a trial, and when a patient drops out of a trial. Information about patient enrollment can become available significantly after we report our expenses for clinical trials, in which case we would change our estimate of the remaining cost of a trial. Costs that are based on clinical data collection and management are recognized based on estimates of unbilled goods and services received. In the event of early termination of a clinical trial, we would accrue an amount based on estimates of the remaining non-cancelable obligations associated with winding down the clinical trial.
Stock Based Compensation
The Financial Accounting Standards Board, or FASB, Statement No. 123 (revised 2004), Share-Based Payment, or SFAS 123R, requires companies to recognize expense in the income statement for the fair value of all share-based payments to employees and directors, including grants of employee stock options and other share based awards. We adopted SFAS 123R on January 1, 2006 using the modified-prospective transition method. We record compensation expense for all awards granted after the date of adoption and for the unvested portion of previously granted awards that were outstanding at the date of adoption. We use the Black-Scholes option valuation model, or Black-Scholes and use the single-option award approach and straight-line attribution method for stock options granted since January 1, 2006. Using this approach, the compensation cost is amortized on a straight-line basis over the vesting period of each respective stock option, generally four years.
We estimate forfeitures when recognizing expense under SFAS 123R and adjust this estimate periodically based on the extent to which future actual forfeitures differ, or are expected to differ, from such estimates. Accordingly, we have estimated forfeiture percentages for the unvested portion of previously granted awards that remain outstanding at the date of adoption and for awards granted subsequent to the date of adoption.
We have granted share-based awards that vest upon achievement of certain performance criteria, or performance awards. The value of these performance awards is the product of the number of shares of our common stock to be issued under the award multiplied by the fair market value of a share of our common stock
46
Table of Contents
PAIN THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
on the date of grant. These performance awards include future performance conditions. We estimate an implicit service period for achieving these performance conditions. Performance awards vest and common stock is issued on achieving performance conditions. We recognize stock-based compensation expense for performance awards when we conclude that achieving a performance condition is probable, consistent with FASB Statement No. 5, Accounting for Contingencies, or SFAS5. We periodically review and update as appropriate our estimates of the implicit service periods and the likelihood of achieving the performance conditions.
Net Income per Share
Basic net income per share is computed on the basis of the weighted-average number of common shares outstanding for the reporting period. Diluted net income per share is computed on the basis of the weighted-average number of common shares outstanding plus dilutive potential common shares outstanding using the treasury-stock method. Potential dilutive common shares consist of outstanding stock options and warrants.
The numerators and denominators in the calculation of basic and diluted net income per share were as follows (in thousands):
| Years Ended December 31, | |||||||||
| 2008 | 2007 | 2006 | |||||||
| Numerators: |
|||||||||
| Net income |
$ | 15,347 | $ | 20,305 | $ | 6,188 | |||
| Denominator |
|||||||||
| Weighted average shares used to compute basic net income per share |
42,252 | 44,150 | 44,146 | ||||||
| Effect of dilutive securities: |
|||||||||
| Dilution from employee stock plans |
1,605 | 1,393 | 1,196 | ||||||
| Dilution from warrants |
— | 133 | 133 | ||||||
| Potential dilutive common shares |
1,605 | 1,526 | 1,329 | ||||||
| Weighted average shares used to compute diluted net income per share |
43,857 | 45,676 | 45,475 | ||||||
| Net income per share: |
|||||||||
| Basic |
$ | 0.36 | $ | 0.46 | $ | 0.14 | |||
| Diluted |
$ | 0.35 | $ | 0.44 | $ | 0.14 | |||
In 2008, 2007, and 2006, we excluded 5.1 million, 3.2 million and 2.2 million shares from diluted net income per share, respectively, as the option exercise price was greater than the average market price per share and the effect would be anti-dilutive.
Comprehensive Income
Comprehensive income is comprised of net income and unrealized holding gains and losses on available-for-sale securities as follows (in thousands):
| Years Ended December 31, | ||||||||||
| 2008 | 2007 | 2006 | ||||||||
| Net income |
$ | 15,347 | $ | 20,305 | $ | 6,188 | ||||
| Other comprehensive income (loss) |
(259 | ) | 956 | 107 | ||||||
| Comprehensive income |
$ | 15,088 | $ | 21,261 | $ | 6,295 | ||||
47
Table of Contents
PAIN THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
Income Taxes
We make estimates and judgments in determining our provision for (benefit from) income taxes. We have accumulated significant deferred tax assets. Deferred income taxes reflect the tax effects of net operating loss and tax credit carryovers and temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Realization of deferred tax assets is dependent upon future earnings, if any. We are uncertain of the timing and amount of any future earnings. Accordingly, except for $1.9 million of deferred tax assets included in other assets as of December 31, 2008, we offset the net deferred tax assets with a valuation allowance. We may in the future determine that more of our deferred tax assets will likely be realized, in which case we will reduce our valuation allowance in the quarter in which such determination is made. If the valuation allowance is reduced, we may recognize a benefit from income taxes in our statement of operations in that period.
3. Collaboration Agreements
King Pharmaceuticals, Inc.
In November 2005, we and King announced a strategic alliance to develop and commercialize Remoxy and other abuse-resistant opioid painkillers. King made an upfront cash payment of $150.0 million to us at the closing of this strategic alliance in December 2005, of which $14.3 million, $23.2 million, and $26.2 million were recorded as program fee revenue for the year ended December 31, 2008, 2007 and 2006, respectively. The strategic alliance provides for us to receive from King up to $150.0 million in milestone payments in the course of clinical development of Remoxy and other abuse-resistant opioid painkillers, of which we received $5.0 million in milestone revenue in the year ended December 31, 2006 and $20.0 million in the year ended December 31, 2008. In addition, subject to certain limitations, King is obligated to fund development expenses incurred by us pursuant to the collaboration agreement, of which $29.4 million, $42.7 million, and $22.7 million was recorded as collaboration revenue for the year ended December 31, 2008, 2007 and 2006 , respectively. King is obligated to fund the commercialization expenses of, and has the exclusive right to market and sell, drugs developed in connection with the strategic alliance. King is obligated to pay us a 20% royalty on net sales of drugs developed in connection with the strategic alliance, except as to the first $1.0 billion in net sales of such drugs, for which the royalty is set at 15%.
Durect Corporation
We have an exclusive, worldwide licensing agreement with Durect Corporation to use a patented technology that forms the basis for a number of oral gel-cap drug candidates, including Remoxy. We have sub-licensed to King certain rights to develop and to commercialize Remoxy and certain other opioid drugs formulated in part with technology we licensed from Durect. Under the agreement with Durect, we control all of the preclinical, clinical, commercial manufacturing and sales/marketing activities for Remoxy and other abuse-resistant opioid painkillers. We reimburse Durect for formulation and related work, and will make milestone payments based on the achievement of certain technical, clinical or regulatory milestones. Durect will supply us with certain components of Remoxy and other abuse-resistant opioid painkillers on a cost-plus basis. We also are responsible to pay Durect royalties on any related drug sales. King is obligated to reimburse us for costs we incur under the agreement with Durect, including royalties.
48
Table of Contents
PAIN THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
4. Cash and Cash Equivalents and Marketable Securities
Cash, cash equivalents and marketable securities held as available-for-sale consisted of the following (in thousands):
| Cash, Cash Equivalents and Marketable Securities | ||||||||||||||||
| Amortized Cost |
Accrued Interest |
Unrealized Gains |
Unrealized Losses |
Estimated Fair Value | ||||||||||||
| December 31, 2008 |
||||||||||||||||
| Cash and cash equivalents: |
||||||||||||||||
| Money market securities |
$ | 153,060 | $ | 98 | $ | — | $ | — | $ | 153,158 | ||||||
| 153,060 | 98 | — | — | 153,158 | ||||||||||||
| Marketable securities available-for-sale: |
||||||||||||||||
| U.S. government and agency obligations |
8,016 | 79 | 233 | — | 8,328 | |||||||||||
| Corporate obligations |
25,348 | 540 | 98 | (1 | ) | 25,985 | ||||||||||
| Asset-backed securities |
2,624 | 5 | — | (5 | ) | 2,624 | ||||||||||
| 35,988 | 624 | 331 | (6 | ) | 36,937 | |||||||||||
| $ | 189,048 | $ | 722 | $ | 331 | $ | (6 | ) | $ | 190,095 | ||||||
| December 31, 2007 |
||||||||||||||||
| Cash and cash equivalents: |
||||||||||||||||
| Money market securities |
$ | 86,344 | $ | 224 | $ | — | $ | (1 | ) | $ | 86,567 | |||||
| 86,344 | 224 | — | (1 | ) | 86,567 | |||||||||||
| Marketable securities available-for-sale: |
||||||||||||||||
| U.S. government and agency obligations |
20,836 | 350 | 133 | — | 21,319 | |||||||||||
| Corporate obligations |
47,760 | 749 | 294 | (12 | ) | 48,791 | ||||||||||
| Asset-backed securities |
48,068 | 156 | 170 | — | 48,394 | |||||||||||
| 116,664 | 1,255 | 597 | (12 | ) | 118,504 | |||||||||||
| $ | 203,008 | $ | 1,479 | $ | 597 | $ | (13 | ) | $ | 205,071 | ||||||
To date we have not recorded any impairment charges on marketable securities related to other-than-temporary declines in market value. We would recognize an impairment charge when the decline in the estimated fair value of a marketable security below the amortized cost is determined to be other-than-temporary. We consider various factors in determining whether to recognize an impairment charge, including the duration of time and the severity to which the fair value has been less than our amortized cost, any adverse changes in the investees’ financial condition and our intent and ability to hold the marketable security for a period of time sufficient to allow for any anticipated recovery in market value.
The gross realized losses and gains on the sale of available-for-sale securities during the years ended December 31, 2008, 2007 and 2006 were not material.
The contractual maturities of our cash equivalents and marketable securities at December 31, 2008 were less than one year.
49
Table of Contents
PAIN THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
The following table presents our assets measured at fair value on a recurring basis at December 31, 2008:
| Level 1 | Level 2 | Total | |||||||
| Money market fund |
$ | — | $ | 153,158 | 153,158 | ||||
| U.S. government and agency obligations |
— | 8,328 | 8,328 | ||||||
| Corporate obligations |
— | 28,609 | 28,609 | ||||||
| $ | — | $ | 190,095 | $ | 190,095 | ||||
5. Property and Equipment
Property and equipment consists of the following (in thousands):
| December 31, | ||||||||
| 2008 | 2007 | |||||||
| Furniture, fixtures and equipment |
$ | 672 | $ | 1,182 | ||||
| Computers and software |
— | 94 | ||||||
| Leasehold improvement |
658 | 2,546 | ||||||
| 1,330 | 3,822 | |||||||
| Accumulated depreciation and amortization |
(556 | ) | (2,215 | ) | ||||
| $ | 774 | $ | 1,607 | |||||
In the year ended December 31, 2008, we wrote-off our property, equipment and leasehold improvements and related accumulated depreciation and amortization related to our lease of office space in South San Francisco because we no longer use that office space. Depreciation and amortization expenses were $452,000, $356,000 and $345,000 in 2008, 2007 and 2006, respectively.
6. Stockholders’ Equity and Stock-Based Compensation
Common Stock
The shelf registration statements we filed with the Securities and Exchange Commission in 2005 and 2003 expired unused in 2008.
Preferred Stock
Our Board of Directors has the authority to issue preferred stock in one or more series and to fix the rights, preferences, privileges, restrictions and the number of shares constituting any series or the designation of the series.
We have a stockholder rights plan designed to guard against partial tender offers and other coercive tactics to gain control of the Company without offering a fair and adequate price and terms to all of the Company’s stockholders. Pursuant to the stockholder rights plan, our Board of Directors declared and paid a dividend of one right to purchase one one-thousandth share of the Company’s Series A Participating Preferred Stock for each outstanding share of our common stock. Each of these rights entitles the registered holder to purchase from us one one-thousandth of a share of Series A Preferred at an exercise price of $40.00, subject to adjustment at any time.
50
Table of Contents
PAIN THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
Stock-Based Compensation
We recognized $10.2 million, $6.3 million and $6.5 million in stock-based compensation expense for the years ended December 31, 2008, 2007 and 2006, respectively. Given our current estimates of future forfeitures, we expect to recognize the compensation cost related to non-vested options as of December 31, 2008 of $14.1 million over the weighted average remaining recognition period of 2.7 years. If all outstanding LTIP awards vest, we would recognize an additional $13.8 million over the remaining recognition period of 3.6 years.
2008 Equity Incentive Plan and 1998 Stock Plan
In 2008, our stockholders approved the 2008 Equity Incentive Plan, or 2008 Equity Plan, to replace the 1998 Stock Plan. The 1998 Stock Plan terminated in 2008. Under the 2008 Equity Plan, our employees, directors and consultants may be granted options that allow for the purchase of shares of our common stock. Incentive stock options may only be granted to employees. Through December 31, 2008 a total of 22,600,000 shares of common stock were authorized for issuance under the 2008 Equity Plan and the 1998 Stock Plan. The 2008 Equity Plan was established with a 10-year duration and terminates in 2018.
Our Board of Directors or a designated Committee of the Board is responsible for administration of the 2008 Equity Plan and the 1998 Stock Plan and determines the terms and conditions of each option granted, consistent with the terms of the plan. Incentive stock options may be granted at a price not less than 100% of the fair market value of the stock on the date of grant (not less than 110% of the fair market value on the date of grant in the case of holders of more than 10% of our voting stock). Options granted under the 2008 Equity Plan and the 1998 Stock Plan generally expire ten years from the date of grant (five years for incentive stock options granted to holders of more than 10% of our voting stock). Forfeited options become available for reissuance under the 2008 Equity Plan.
The 2008 Equity Plan also provides for the automatic grant of options to purchase shares of common stock to outside directors. On the date of each annual stockholders’ meeting, each outside director is automatically granted an option to purchase 25,000 shares of common stock. The term of the option is ten years, the exercise price is 100% of the fair market value of the stock on the date of grant, and the option becomes exercisable as to 25% of the shares on the anniversary of its date of grant provided the optionee continues to serve as a director on such dates.
51
Table of Contents
PAIN THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
The following summarizes stock option activity for the years ended December 31, 2008, 2007 and 2006:
| Number of Options |
Weighted Average Exercise Price |
Weighted Average Remaining Contractual Term |
Aggregate Intrinsic Value | ||||||||
| In years | In millions | ||||||||||
| Options outstanding as of December 31, 2005 |
6,993,492 | $ | 6.74 | ||||||||
| Granted |
1,617,200 | $ | 8.35 | ||||||||
| Exercised |
(304,180 | ) | $ | 4.31 | |||||||
| Forfeited |
(93,527 | ) | $ | 8.56 | |||||||
| Options outstanding as of December 31, 2006 |
8,212,985 | $ | 7.12 | ||||||||
| Granted |
2,263,400 | $ | 9.01 | ||||||||
| Exercised |
(400,442 | ) | $ | 5.70 | |||||||
| Forfeited |
(78,197 | ) | $ | 8.06 | |||||||
| Options outstanding as of December 31, 2007 |
9,997,746 | $ | 7.60 | 6.93 | $ | 30.9 | |||||
| Granted |
1,835,600 | $ | 7.37 | ||||||||
| Exercised |
(284,308 | ) | $ | 6.44 | |||||||
| Forfeited |
(91,223 | ) | $ | 8.64 | |||||||
| Options outstanding as of December 31, 2008 |
11,457,815 | $ | 7.58 | 6.52 | $ | 1.8 | |||||
| Vested and expected to vest at December 31, 2008 |
11,188,544 | $ | 7.57 | 6.46 | $ | 1.8 | |||||
| Exercisable at December 31, 2008 |
7,484,079 | $ | 7.33 | 5.35 | $ | 1.7 | |||||
The pre-tax intrinsic value of options exercised was approximately $4.1 million in 2008, $1.6 million in 2007 and $1.5 million in 2006, calculated by multiplying options exercised each year by the difference between our stock price on the date of exercise and the exercise price of the options. Shares reserved for issuance and available for grant under the 2008 Equity Incentive Plan were 5,868,485 as of December 31, 2008 and 1,488,436 under our 1998 Stock Plan as of December and 2007.
The following summarizes information about stock options outstanding at December 31, 2008:
| Options outstanding | Options exercisable | |||||||||||
| Range of exercise prices |
Number of options |
Weighted average remaining contractual life (in years) |
Weighted average exercise price |
Number of vested options |
Weighted average exercise price | |||||||
| $ 0.10—$ 6.71 |
2,735,709 | 5.71 | $ | 5.52 | 2,167,853 | $ | 5.37 | |||||
| 6.82— 7.51 |
3,095,013 | 6.33 | 7.23 | 1,990,253 | 7.09 | |||||||
| 7.60— 8.25 |
2,767,144 | 7.10 | 8.04 | 1,702,747 | 7.94 | |||||||
| 8.32— 10.53 |
2,704,949 | 7.23 | 9.10 | 1,468,226 | 8.90 | |||||||
| 14.13— 18.63 |
155,000 | 1.84 | 16.30 | 155,000 | 16.30 | |||||||
| $ 0.10—$18.63 |
11,457,815 | 6.52 | $ | 7.58 | 7,484,079 | $ | 7.33 | |||||
52
Table of Contents
PAIN THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
Determining the Fair Value of Options
We use Black-Scholes to estimate the fair value of options granted. Black-Scholes considers a number of factors, including the market price and volatility of our common stock. We used the following forward-looking range of assumptions to value each stock option granted to employees and directors during the years ended December 31, 2008, 2007 and 2006:
| 2008 | 2007 | 2006 | ||||
| Volatility |
48% to 54% | 60% to 67% | 68% to 74% | |||
| Risk-free interest rates |
2% to 4% | 4% to 5% | 5% | |||
| Expected life of option |
5 years | 5 years | 5 years | |||
| Dividend yield |
— | — | — |
Our volatility assumption is based on reviews of the historical volatility of our common stock. Our risk-free interest rate assumption is based on yields of US treasury notes in effect at the date of grant. Our expected life of options granted assumption is based on actual historical option exercises. Our dividend yield assumption is based on the fact that we have never paid cash dividends and do not anticipate paying cash dividends in the foreseeable future. Using Black-Scholes and these factors, the weighted average fair value of stock options granted to employees and directors was $7.37 for the year ended December 31, 2008 and $5.16 for the years ended December 31, 2007 and 2006. In 2006, 2007 and 2008 we used an expected forfeiture rate of 5%. Our estimated expected forfeiture rate is based on historical cancellations of options.
We estimate the fair value of stock options granted to non-employees using forward-looking assumptions similar to those used for stock options granted to employees and directors and appropriate for the terms underlying the stock options granted to non-employees. We re-measure the compensation expense for options granted to non-employees over the related vesting period. The expense related to stock options granted to non-employees was approximately $0.1 million, $0.4 million, and $0.2 million for the years ended December 31, 2008, 2007 and 2006, respectively.
Performance Awards
We granted performance awards in 2008 for 2.0 million shares. The total value of these performance awards was $17.1 million. In connection with FDA acceptance for filing of the NDA for Remoxy, 0.4 million of these performance awards vested. We recognized related stock-based compensation expense of $1.7 million in research and development expenses and $1.6 million in general and administrative expenses. If the remaining 1.6 million shares of performance awards do not vest within four years of the date of grant, the awards expire and the underlying shares are returned to the 2008 Equity Incentive Plan.
2000 Employee Stock Purchase Plan
Under the 2000 Employee Stock Purchase Plan, or the Purchase Plan, eligible employees may purchase common stock through payroll deductions of up to 15% of the employee’s compensation. The purchase price of the stock is generally 85% of the lower of the fair market value of the common stock at the beginning of the offering period or at the end of the purchase period. A total of 500,000 shares of common stock have been reserved for issuance under the Purchase Plan. Shares reserved for issuance under the Purchase Plan may be automatically increased each year by the amount equal to the lesser of (i) 500,000 shares, (ii) 1% of the initially outstanding shares of common stock on such date, or (iii) an amount determined by our Board of Directors. We have issued 494,957 shares of common stock pursuant to the Purchase Plan through December 31, 2008, leaving 255,043 shares reserved for issuance.
53
Table of Contents
PAIN THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
The weighted-average fair value of purchase rights granted was $6.31, $6.62, and $2.15 in 2008, 2007 and 2006, respectively, calculated using Black-Scholes with an expected life of 1 year in 2008, 2007 and 2006 with no dividend yield. We assumed volatility was 40% to 49% in 2008, 34% in 2007, and 55% in 2006. We used risk free interest rates of 1% to 2% in 2008, 5% in 2007, and 5% in 2006. The expense related to the Employee Stock Purchase Plan was $0.1 million, $0.1 million and $0.2 million for the years ended December 31, 2008, 2007 and 2006, respectively.
Stock Repurchase Program
Under a stock repurchase program approved by our Board of Directors in March 2007 and extended in March 2008, we completed the repurchase of $30.0 million of our common stock during the year ended December 31, 2008. We used the par value method of accounting for our stock repurchases. The excess of the cost of the shares acquired over the par value is allocated to additional paid-in capital based on the weighted average sales price per issued share with the remainder charged to accumulated deficit.
Warrants
As of December 31, 2008, we have no outstanding exercisable warrants.
7. Employee 401(k) Benefit Plan
We have a defined-contribution savings plan under Section 401(k) of the Internal Revenue Code. The plan covers substantially all employees. Employees are eligible to participate in the plan the first day of the month after hire and may contribute up to the current statutory limits under Internal Revenue Service regulations. The 401(k) plan permits us to make additional matching contributions on behalf of all employees. Through December 31, 2008, we have not made any matching contributions.
8. Accounting for Sabbatical Leave
On January 1, 2007 we adopted EITF Issue No. 06-2, “Accounting for Sabbatical Leave and Other Similar Benefits Pursuant to FASB Statement No. 43, Accounting for Compensated Absences,” and recorded a one-time charge of $0.4 million to accumulated deficit for sabbatical leave as a cumulative effect of a change in accounting principle. Our expense for sabbatical leave was immaterial for the year ended December 31, 2007 and 2008.
9. Income Taxes
We had taxable income for 2008 primarily due to the combination of milestone payments we received from King and interest income. We reduced our expected tax for 2008 with a combination of net operating losses and tax credits earned from prior years.
In 2005, King made an upfront cash payment of $150.0 million to us in connection with our strategic alliance. We recognized as revenue for tax purposes $3.7 million and $146.3 million of the upfront cash payment in 2005 and 2006, respectively. We had taxable income for 2006 primarily due to the recognition in 2006 of the $146.3 million of the upfront cash payment. We reduced our expected tax for 2006 with deductions related to a combination of our net operating losses and tax credits from prior years. While we continued to recognize program fee revenue in our income statement for 2008 and 2007 for the recognition of the upfront cash payment, this program fee revenue is excluded from our taxable income for 2008 and 2007. This exclusion combined with other differences between our income before income taxes and our taxable income results in no taxable income for 2007 and no related provision for income taxes for 2007. All of our net income is domestic.
54
Table of Contents
PAIN THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
Uncertainty in Income Taxes
We adopted the provisions of FASB Interpretation No. 48, “Accounting for Uncertainty in Income Taxes,” or Interpretation 48, on January 1, 2007. As a result of the implementation of Interpretation 48, we adjusted the balance sheet as of January 1, 2007 by recording a non-current liability of $0.8 million and a non-current deferred tax asset of $0.7 million. The non-current liability provides a source of future taxable income against which the non-current deferred tax asset of $0.7 million was recognized without offset by a valuation allowance.
As of the adoption date, we had unrecognized tax benefits of approximately $3.5 million related primarily to tax credits. A reconciliation of the beginning and ending amount of liability recorded for 2008 and 2007 follows (in thousands):
| 2008 | 2007 | |||||
| Beginning Balance |
$ | 3,800 | $ | 3,500 | ||
| Additions based on tax positions related to the current year |
400 | 300 | ||||
| Additions (reductions) for tax positions of prior years |
— | — | ||||
| Reductions for tax positions of prior years |
— | — | ||||
| Settlements |
— | — | ||||
| Lapse of applicable statute of limitations |
— | — | ||||
| Ending Balance |
$ | 4,200 | $ | 3,800 | ||
The total amount of unrecognized tax benefit that, if recognized, would benefit our effective tax rate, is $67 thousand. Because of net operating loss and research credit carryforwards, substantially all of our tax years, from 1998 through 2008, remain open to U.S. federal and California state tax examinations.
We classify interest and penalties recognized pursuant to Interpretation 48 as interest expense. There were no interest or penalties related to unrecognized tax benefits accrued at December 31, 2006. Interest expense and penalties related to unrecognized tax benefits were immaterial for the years ended December 31, 2008 and 2007.
Provision for (benefit from) income taxes
In 2008 we had an income tax benefit of $0.6 million comprised of a federal tax benefit of $0.8 million, net of a provision for state taxes of $0.2 million. The federal income tax benefit is primarily due to the recognition in 2008 of previously generated net operating losses and tax credits. We had $1.4 million of deferred benefits and $0.8 million in current tax expense in our benefit from income taxes in 2008. We did not have a provision for income taxes in 2007. Our provision for income taxes for 2006 was $2.9 million, of which $2.7 million was for federal tax and $0.2 million was for state tax. A reconciliation between our provision for (benefit from) income taxes for 2008 and 2006 and the amounts computed by multiplying income before provision for (benefit from) income tax by the U.S. statutory tax rate follows (in thousands):
| 2008 | 2006 | |||||||
| Tax at U.S. statutory tax rate of 35% |
$ | 4,968 | $ | 3,190 | ||||
| State taxes |
180 | 188 | ||||||
| Research credits |
(415 | ) | (481 | ) | ||||
| Equity-based compensation |
579 | 699 | ||||||
| Change in valuation allowance |
(5,935 | ) | (679 | ) | ||||
| Other |
6 | 10 | ||||||
| Provision for income taxes |
$ | (617 | ) | $ | 2,927 | |||
55
Table of Contents
PAIN THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
Deferred tax assets and valuation allowance
Deferred tax assets reflect the tax effects of net operating loss and tax credit carryovers and temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of our deferred tax assets are as follows (in thousands):
| December 31, | ||||||||||||
| 2008 | 2007 | 2006 | ||||||||||
| Deferred tax assets: |
||||||||||||
| Net operating loss carryforwards |
$ | 100 | $ | 900 | $ | — | ||||||
| Deferred license fee revenue |
32,900 | 39,400 | 48,800 | |||||||||
| Research & development credits |
3,400 | 3,700 | 3,100 | |||||||||
| Stock-related compensation |
5,700 | 4,000 | 2,500 | |||||||||
| Other |
1,400 | 1,600 | 2,900 | |||||||||
| Total deferred tax assets |
43,500 | 49,600 | 57,300 | |||||||||
| Valuation allowance |
(41,600 | ) | (49,100 | ) | (57,300 | ) | ||||||
| Net deferred tax assets |
$ | 1,900 | $ | 500 | $ | — | ||||||
Except for deferred tax assets recognized pursuant to Interpretation 48, realization of deferred tax assets is dependent upon future earnings, if any, the timing and amount of which are uncertain. We have concluded that it is more likely than not that these deferred tax assets will not be realized. Accordingly, these deferred tax assets have been offset by a valuation allowance. We will continue to assess whether or not our deferred tax assets will be realized based on actual and forecasted operating results. The net operating loss carryforwards from 2007 expire in 2027. Approximately $0.3 million of the valuation allowance at December 31, 2008 relates to the tax benefits associated with excess tax deductions resulting from stock option transactions. This amount will be credited to additional paid-in capital when realized in a reduction to income taxes payable. The valuation allowance decreased by $7.5 million, $8.2 million and $4.4 million in 2008, 2007 and 2006, respectively.
As of December 31, 2008, we had federal research and development tax credits of approximately $6.1 million, which expire in the years 2018 through 2027 and state research and development tax credits of approximately $0.9 million.
10. Leases and Commitments
We conduct our product research and development programs through a combination of internal and collaborative programs that include, among others, arrangements with universities, contract research organizations and clinical research sites. We have contractual arrangements with these organizations, however these contracts are cancelable on thirty days notice and our obligations under these contracts are largely based on services performed.
We currently lease approximately 41,200 square feet of office space pursuant to non-cancelable operating leases that will expire in 2010 and 2012. Future minimum lease payments for our leases are as follows for the years ended December 31, (in thousands):
| 2009 | 2010 | 2011 | 2012 | Total | |||||||||||
| Future minimum lease payments |
$ | 743 | $ | 713 | $ | 570 | $ | 339 | $ | 2,365 | |||||
56
Table of Contents
PAIN THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
Rent expense was $702,000, $501,000, and $178,000 for the years ended December 31, 2008, 2007, and 2006 respectively.
11. Selected Quarterly Financial Data (Unaudited) (in thousands except per share data)
| Quarters Ended | ||||||||||||||
| March 31 | June 30 | September 30 | December 31 | |||||||||||
| 2008 |
||||||||||||||
| Total revenue |
$ | 14,639 | $ | 10,547 | $ | 30,294 | $ | 8,245 | ||||||
| Net income (loss) |
$ | 2,570 | $ | (1,025 | ) | $ | 15,191 | $ | (1,389 | ) | ||||
| Basic income (loss) per share |
$ | 0.06 | $ | (0.02 | ) | $ | 0.37 | $ | 0.03 | |||||
| Diluted income (loss) per share |
$ | 0.06 | $ | (0.02 | ) | $ | 0.35 | $ | 0.03 | |||||
| 2007 |
||||||||||||||
| Total revenue |
$ | 22,054 | $ | 14,065 | $ | 15,810 | $ | 14,055 | ||||||
| Net income |
$ | 12,636 | $ | 3,365 | $ | 3,173 | $ | 1,131 | ||||||
| Basic income per share |
$ | 0.28 | $ | 0.08 | $ | 0.07 | $ | 0.03 | ||||||
| Diluted income per share |
$ | 0.28 | $ | 0.07 | $ | 0.07 | $ | 0.02 | ||||||
57
Table of Contents
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
None.
| Item 9A. | Controls and Procedures |
Evaluation of disclosure controls and procedures. Our management evaluated, with the participation of our Chief Executive Officer and our Chief Financial Officer, the effectiveness of our disclosure controls and procedures as of the end of the period covered by this Annual Report on Form 10-K. Based on this evaluation, our Chief Executive Officer and our Chief Financial Officer have concluded that our disclosure controls and procedures are effective to ensure that information we are required to disclose in reports that we file or submit under the Securities Exchange Act of 1934 is recorded, processed, summarized and reported within the time periods specified in Securities and Exchange Commission rules and forms.
Management’s annual report on internal control over financial reporting. We are responsible for establishing and maintaining adequate internal control over our financial reporting. We have assessed the effectiveness of internal control over financial reporting as of December 31, 2008. Our assessment was based on criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission, or COSO, in Internal Control—Integrated Framework.
Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Our internal control over financial reporting includes those policies and procedures that:
| (1) | pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect our transactions and dispositions of our assets; |
| (2) | provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and board of directors; and |
| (3) | provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of our assets that could have a material effect on the financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Based on using the COSO criteria, we believe our internal control over financial reporting as of December 31, 2008 was effective.
Changes in internal control over financial reporting. There was no change in our internal control over financial reporting that occurred during the quarter ended December 31, 2008 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Our independent registered public accounting firm, Ernst & Young LLP, has audited the financial statements included in this Annual Report on Form 10-K and has issued a report on the effectiveness of our internal control over financial reporting. The attestation report of Ernst & Young LLP is included below.
58
Table of Contents
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders
Pain Therapeutics, Inc.
We have audited Pain Therapeutics, Inc.’s internal control over financial reporting as of December 31, 2008, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). Pain Therapeutics, Inc.’s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s annual report on internal control over financial reporting. Our responsibility is to express an opinion on the company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A Company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Pain Therapeutics, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2008, based on the COSO criteria.
ERNST & YOUNG LLP
San Jose, California
February 11, 2009
59
Table of Contents
| Item 9B. | Other Information |
None.
| Item 10. | Directors and Executive Officers and Corporate Governance |
The information regarding our directors, executive officers and the audit committee of our board of directors is incorporated by reference from “Directors and Executive Officers” in our Proxy Statement for our 2009 Annual Meeting of Stockholders.
Section 16(a) Beneficial Ownership Reporting Compliance
Section 16(a) of the Securities Exchange Act of 1934, as amended requires our executive officers and directors and persons who own more than ten percent (10%) of a registered class of our equity securities to file reports of ownership and changes in ownership with the Securities and Exchange Commission, or SEC. Executive officers, directors and greater than ten percent (10%) stockholders are required by Commission regulation to furnish us with copies of all Section 16(a) forms they file. We believe all of our executive officers and directors complied with all applicable filing requirements during the fiscal year ended December 31, 2008.
Code of Ethics
We have adopted a Code of Ethics that applies to all of our directors, officers and employees, including our principal executive officer, principal financial officer and principal accounting officer. We publicize the Code of Ethics through posting the policy on our website, http://www.paintrials.com. We will disclose on our website any waivers of, or amendments to, our Code of Ethics.
| Item 11. | Executive Compensation |
The information required by this Item is incorporated by reference from our definitive Proxy Statement referred to in Item 10 above where it appears under the heading “Executive Compensation and Other Matters.”
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
The information required by this Item regarding security ownership of certain beneficial owners and management is incorporated by reference from our definitive Proxy Statement referred to in Item 10 above where it appears under the heading “Security Ownership of Certain Beneficial Owners and Management.”
The following table summarizes the securities authorized for issuance under our equity compensation plans as of December 31, 2008:
| Number of Securities to be Issued Upon Exercise of Outstanding Options, Warrants and Rights |
Weighted Average Exercise Price of Outstanding Options, Warrants and Rights |
Number of Securities Remaining Available for Future Issuance Under Equity Compensation Plans | |||||
| Equity compensation plans approved by stockholders |
12,932,815 | $ | 7.58 | 6,123,528 | |||
| Equity compensation plans not approved by stockholders |
— | — | — | ||||
| Total |
12,932,815 | $ | 7.58 | 6,123,528 | |||
60
Table of Contents
| Item 13. | Certain Relationships and Related Transactions and Director Independence |
The information required by this Item is incorporated by reference from our definitive Proxy Statement referred to in Item 10 above where it appears under the heading “Certain Relationships and Related Transactions.”
| Item 14. | Principal Accountant Fees and Services |
The information required by this Item is incorporated by reference from our definitive Proxy Statement referred to in Item 10 above where it appears under the heading “Principal Accountant Fees and Services.”
| Item 15. | Exhibits and Financial Statement Schedules |
| (a) | The following documents are filed as part of this Form 10-K: |
| (1) | Financial Statements (included in Part II of this report): |
Reports of Independent Registered Public Accounting Firm
Balance Sheets
Statements of Operations
Statement of Stockholders’ Equity
Statements of Cash Flows
Notes to Financial Statements
| (2) | Financial Statement Schedules: |
All financial statement schedules are omitted because the information is inapplicable or presented in the notes to the financial statements.
| (3) | Exhibits: |
| Exhibit Number |
Description of Document | |
| 3.1(1) | Amended and Restated Certificate of Incorporation. | |
| 3.2(2) | Amended and Restated Bylaws. | |
| 4.1(1) | Specimen Common Stock Certificate. | |
| 4.2(3) | Preferred Stock Rights Agreement, dated as of April 28, 2005, between the Company and Mellon Investor Services LLC, including the Certificate of Designation, the form of Rights Certificate and Summary of Rights attached thereto as Exhibits A, B and C, respectively. | |
| 10.1(4) | Form of Indemnification Agreement between Pain Therapeutics and each of its directors and officers. | |
| 10.2(4) | 1998 Equity Incentive Plan and form of agreements thereunder. | |
| 10.3(4) | 2000 Employee Stock Purchase Plan and form of agreements thereunder. | |
| 10.4(5) | Employment Agreement dated October 23, 2001, between Registrant and Nadav Friedmann, PhD. M.D. | |
| 10.5(6) | Lease Agreement dated July 21, 2000 between Registrant and Goss-Jewett Company of Northern California. | |
| 10.6(7) | Collaboration Agreement dated November 9, 2005, between Registrant and King Pharmaceuticals, Inc. | |
61
Table of Contents
| Exhibit Number |
Description of Document | |
| 10.7(7) | License Agreement dated November 9, 2005, between Registrant and King Pharmaceuticals, Inc. | |
| 10.8(7) | Development and License Agreement dated December 19, 2002 between Registrant and DURECT Corporation and Southern Biosystems, Inc. | |
| 10.9(7) | Amendment dated December 15, 2005 to Development and License Agreement dated December 19, 2002 between Registrant and DURECT Corporation and Southern Biosystems, Inc. | |
| 10.10(8) | Sublease Agreement, dated as of July 17, 2007, between Registrant and Oracle USA, Inc. | |
| 10.11(9) | 2008 Equity Incentive Plan and form of agreements thereunder. | |
| 10.12 | Employment Agreement dated July 1, 1998 and amended December 17, 2008 between Registrant and Remi Barbier. | |
| 10.13 | Employment Agreement dated August 29, 2000 and amended December 30, 2008 between Registrant and Grant L. Schoenhard, Ph.D. | |
| 10.14 | Employment Agreement dated November 18, 2002 and amended December 30, 2008 between Registrant and Peter S. Roddy. | |
| 23.1 | Consent of Independent Registered Public Accounting Firm. | |
| 24.1 | Power of Attorney (see page 63). | |
| 31.1 | Certification of Principal Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 31.2 | Certification of Principal Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 32.1 | Certifications of the Chief Executive Officer and the Chief Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| (1) | Incorporated by reference from exhibits to our report on Form 10-Q for the period ending June 30, 2005. |
| (2) | Incorporated by reference from exhibits to our report on Form 10-Q for the period ending March 31, 2005. |
| (3) | Incorporated by reference from exhibits to our report on Form 8-K as filed with the Securities and Exchange Commission on May 3, 2005. |
| (4) | Incorporated by reference from our registration statement on Form S-1, registration number 333-32370, declared effective by the Securities and Exchange Commission on July 13, 2000. |
| (5) | Incorporated by reference from exhibits to our report on Form 10-K for the period ending December 31, 2001. |
| (6) | Incorporated by reference from exhibits to our report on Form 10-Q for the period ending September 30, 2000. |
| (7) | Incorporated by reference from exhibits to our report on Form 10-K for the period ending December 31, 2005. |
| (8) | Incorporated by reference from exhibits to our report on Form 10-Q for the period ending September 30, 2007. |
| (9) | Incorporated by reference from exhibits to our report on Form 8-K as filed with the Securities and Exchange Commission on May 29, 2008. |
| (b) | Exhibits |
The exhibits listed under Item 15(a)(3) hereof are filed as part of this Form 10-K other than Exhibit 32.1, which shall be deemed furnished.
| (c) | Financial Statement Schedules |
All financial statement schedules are omitted because the information is inapplicable or presented in the notes to the financial statements.
62
Table of Contents
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| PAIN THERAPEUTICS, INC. | ||
| By: |
/s/ REMI BARBIER | |
| Remi Barbier | ||
| President, Chief Executive Officer and Chairman of the Board of Directors | ||
Dated: February 12, 2009
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Remi Barbier and Peter S. Roddy, and each of them, his true and lawful attorneys-in-fact, each with full power of substitution, for him in any and all capacities, to sign any amendments to this Annual Report on Form 10-K and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that each of said attorneys-in-fact or their substitute or substitutes may do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature |
Title |
Date | ||
| /S/ REMI BARBIER Remi Barbier |
President, Chief Executive Officer and Chairman of the Board of Directors (Principal Executive Officer) |
February 12, 2009 | ||
| /S/ PETER S. RODDY Peter S. Roddy |
Vice President and Chief Financial Officer (Principal Financial and Accounting Officer) |
February 12, 2009 | ||
| /S/ NADAV FRIEDMANN, PH.D., M.D. Nadav Friedmann, Ph.D., M.D. |
Chief Operating and Medical |
February 12, 2009 | ||
| /S/ PATRICK SCANNON, M.D, PH.D. Patrick Scannon, M.D., Ph.D. |
Director |
February 12, 2009 | ||
| /S/ ROBERT Z. GUSSIN, PH.D. Robert Z. Gussin, Ph.D. |
Director |
February 12, 2009 | ||
| /S/ MICHAEL J. O’DONNELL, ESQ. Michael J. O’Donnell, Esq. |
Director and Secretary |
February 12, 2009 | ||
| /S/ SANFORD R. ROBERTSON Sanford R. Robertson |
Director |
February 12, 2009 | ||
63
Table of Contents
EXHIBIT INDEX
| Exhibit Number |
Description of Document | |
| 3.1(1) | Amended and Restated Certificate of Incorporation. | |
| 3.2(2) | Amended and Restated Bylaws. | |
| 4.1(1) | Specimen Common Stock Certificate. | |
| 4.2(3) | Preferred Stock Rights Agreement, dated as of April 28, 2005, between the Company and Mellon Investor Services LLC, including the Certificate of Designation, the form of Rights Certificate and Summary of Rights attached thereto as Exhibits A, B and C, respectively. | |
| 10.1(4) | Form of Indemnification Agreement between Pain Therapeutics and each of its directors and officers. | |
| 10.2(4) | 1998 Equity Incentive Plan and form of agreements thereunder. | |
| 10.3(4) | 2000 Employee Stock Purchase Plan and form of agreements thereunder. | |
| 10.4(5) | Employment Agreement dated October 23, 2001, between Registrant and Nadav Friedmann, PhD. M.D. | |
| 10.5(6) | Lease Agreement dated July 21, 2000 between Registrant and Goss-Jewett Company of Northern California. | |
| 10.6(7) | Collaboration Agreement dated November 9, 2005, between Registrant and King Pharmaceuticals, Inc. | |
| 10.7(7) | License Agreement dated November 9, 2005, between Registrant and King Pharmaceuticals, Inc. | |
| 10.8(7) | Development and License Agreement dated December 19, 2002 between Registrant and DURECT Corporation and Southern Biosystems, Inc. | |
| 10.9(7) | Amendment dated December 15, 2005 to Development and License Agreement dated December 19, 2002 between Registrant and DURECT Corporation and Southern Biosystems, Inc. | |
| 10.10(8) | Sublease Agreement, dated as of July 17, 2007, between Registrant and Oracle USA, Inc. | |
| 10.11(9) | 2008 Equity Incentive Plan and form of agreements thereunder. | |
| 10.12 | Employment Agreement dated July 1, 1998 and amended December 17, 2008 between Registrant and Remi Barbier. | |
| 10.13 | Employment Agreement dated August 29, 2000 and amended December 30, 2008 between Registrant and Grant L. Schoenhard, Ph.D. | |
| 10.14 | Employment Agreement dated November 18, 2002 and amended December 30, 2008 between Registrant and Peter S. Roddy. | |
| 23.1 | Consent of Independent Registered Public Accounting Firm. | |
| 24.1 | Power of Attorney (see page 63). | |
| 31.1 | Certification of Principal Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 31.2 | Certification of Principal Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 32.1 | Certifications of the Chief Executive Officer and the Chief Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| (1) | Incorporated by reference from exhibits to our report on Form 10-Q for the period ending June 30, 2005. |
| (2) | Incorporated by reference from exhibits to our report on Form 10-Q for the period ending March 31, 2005. |
| (3) | Incorporated by reference from exhibits to our report on Form 8-K as filed with the Securities and Exchange Commission on May 3, 2005. |
| (4) | Incorporated by reference from our registration statement on Form S-1, registration number 333-32370, declared effective by the Securities and Exchange Commission on July 13, 2000. |
64
Table of Contents
| (5) | Incorporated by reference from exhibits to our report on Form 10-K for the period ending December 31, 2001. |
| (6) | Incorporated by reference from exhibits to our report on Form 10-Q for the period ending September 30, 2000. |
| (7) | Incorporated by reference from exhibits to our report on Form 10-K for the period ending December 31, 2005. |
| (8) | Incorporated by reference from exhibits to our report on Form 10-Q for the period ending September 30, 2007. |
| (9) | Incorporated by reference from exhibits to our report on Form 8-K as filed with the Securities and Exchange Commission on May 29, 2008. |
65
EXHIBIT 10.12
PAIN THERAPEUTICS, INC.
EMPLOYMENT AGREEMENT
This Employment Agreement (the “Agreement”) is made by and between Pain Therapeutics, Inc., a Delaware corporation (the “Company”) and Remi Barbier (“Executive”) as of July 1, 1998.
RECITALS
A. The Company desires to have Executive’s active services as President, Chief Executive Officer and Chairman of the Board of the Company for the period set forth in this Agreement.
B. The Company and Executive desire to enter into this Agreement on the terms and conditions set forth in this Agreement.
AGREEMENT
NOW, THEREFORE, in consideration of the mutual covenants herein contained, and in consideration of Executive’s continued employment by the Company, the parties hereto agree as follows:
1. Duties and Scope of Employment
(a) Duties. The Company shall employ Executive to render services to the Company. Executive agrees that he will devote his full business time and efforts to the business of the Company, excluding up to four (4) weeks paid vacation per year, and, in addition, sick leave in accordance with the Company’s policies. In the course of Executive’s employment, Executive shall perform the duties of President and Chief Executive Officer of the Company under the direction of the Board of Directors.
(b) Term of Employment. Executive’s employment with the Company pursuant to this Agreement shall commence on the date hereof and shall continue until thirty-six (36) months thereafter (the “Employment Period”), provided that such term shall automatically renew for an additional 12 months at the end of such initial term and each subsequent renewal, unless ninety (90) days prior to the date of such renewal either party shall give written notice to the other party of cancellation, and in such event the Executive’s employment hereunder shall terminate. Subject to Executive’s right to severance compensation under certain circumstances as set forth herein, the employment relation of Executive with the Company shall be terminable upon sixty (60) days prior written notice by either party.
(c) Director. As long as Executive serves as President and Chief Executive Officer, Executive shall be nominated to serve on the Company’s Board of Directors. Executive agrees to submit his resignation immediately as a director if Executive ceases to be President and Chief Executive Officer, unless removed as President and Chief Executive Officer without Cause as defined below.
2. Compensation
(a) Base Compensation. The Company shall pay the Executive as compensation for his service a base salary at the annualized rate of Two Hundred Fifty Thousand Dollars ($250,000) (“Salary”). Such Salary shall increase each anniversary date by the greater of the Company’s Consumer Price Index or five percent (5%), whichever is greater, and shall be reviewed at least annually and may be increased from time to time subject to accomplishment of such performance and contribution goals and objectives as may be established and agreed upon from time to time by the Board of Directors and the Executive. Such Salary shall be paid periodically in accordance with normal Company payroll. The annual compensation (including bonus and benefit amounts pursuant to Section 2(b) and (c) below) specified in this Section 2(a), together with any increases in such compensation that the Board of Directors may grant from time to time, is referred to in this Agreement as “Base Compensation.”
(b) Deferment of Salary. One Hundred Thousand Dollars ($100,000) of the Executive’s annual Salary shall be deferred until such time as the Company raises a total of Four Million Dollars ($4,000,000) or more in any combination of debt or equity capital from outside investors, corporate partners, alliances, mergers, etc, The Executive shall automatically forgive the Company any deferred compensation in month 36 of this Employment Agreement, provided Executive is still an employee in good standing at such time. Any deferred compensation shall be immediately due upon termination of the Executive’s employment without Cause (as defined below).
(c) Bonuses. Beginning with the Company’s 1998 fiscal year and for each fiscal year thereafter during the Employment Period, the Executive will be eligible to receive an annual bonus based upon certain criteria to be agreed upon by Executive and the Board of Directors to be paid on January 15 of each calendar year hereafter, provided that Executive is employed by the Company on such dates.
(d) Executive Benefits. The Executive shall be eligible to participate in the employee benefit plans and executive compensation programs maintained by the Company applicable to other key executives of the Company, including (without limitation) retirement plans, savings or profit-sharing plans, deferred compensation plans, supplemental retirement or excess benefit plans, life, disability, health, accident and other insurance programs, paid vacations, and similar plans or programs, subject in each case to the generally applicable terms and conditions of the plan or program in question and to the determination of any committee administering such plan or program.
(e) Expenses During Employment. The Company shall reimburse the Executive for all reasonable business, entertainment and travel expenses actually incurred or paid by the Executive in the performance of his services on behalf of the Company, in accordance with the Company’s expense reimbursement policy as from time to time in effect.
3. Termination of Employment
(a) By Death. The Employment Period shall terminate automatically upon the death of the Executive. In such event, the Company shall pay to Executive’s beneficiaries or his estate, as the case may be, any accrued Salary, the pro rata amount of the guaranteed annual bonus, any vested deferred compensation (other than pension plan or profit-sharing plan benefits which will be paid in accordance with the applicable plan), any benefits under any plan of the Company in which Executive is a participant to the full extent of Executive’s rights under such plan, any accrued vacation pay and any appropriate business expenses incurred by Executive in connection with his duties hereunder, all to the date of termination (collectively “Accrued Compensation”), but no other compensation or reimbursement of any kind, including, without limitation, severance compensation, and thereafter, the Company’s obligations hereunder shall terminate. Nothing in this Section shall affect any entitlement of the Executive’s heirs to the benefits of any life insurance provided by the Company as set forth above.
(b) By Disability. If the Executive is prevented from properly performing his duties hereunder by reason of any physical or mental incapacity for a period of more than sixty (60) days in the aggregate in any 365-day period, then, to the extent permitted by law, the Company may terminate the Employment Period on the 60th day of such incapacity. In such event, the Company shall pay to Executive all Accrued Compensation, and shall continue to pay to Executive the Salary and the pro rata amount of the guaranteed annual bonus until such time as Executive shall become entitled to receive disability insurance payments under the disability insurance policy maintained by the Company (but not more than ninety (90) days following termination), but no other compensation or reimbursement of any kind, including without limitation, severance compensation, and thereafter the Company’s obligations hereunder shall terminate. Nothing in this Section shall affect Executive’s rights under any disability plan in which he is a participant.
(c) By Resignation or By Company for Cause. If Executive’s employment with the, Company terminates due to his voluntary resignation or if the Company terminates Executive’s employment due to Cause (as defined below), the Company shall pay Executive all Accrued Compensation less the pro rata, amount of the annual guaranteed bonus (which shall not be paid to Executive), but no other compensation or reimbursement of any End, including without limitation, severance compensation, and thereafter the Company’s obligations hereunder shall terminate. Termination shall be for “Cause” in the event of the occurrence of any of the following: (i) any intentional action or intentional failure to act that was performed in bad faith and to the detriment of the Company; (ii) any intentional refusal or intentional failure to act in accordance with any lawful and proper direction or order of the Board; (iii) any willful and habitual neglect of the duties of full or part-time employment as assigned by the Board from time to time; or (iv) any conviction of a felony crime under the state or federal laws of the United States of America; provided that, in the event that any of the foregoing events is capable of being cured, the Company shall provide written notice to the Purchaser describing the nature of such event, and the Purchaser shall thereafter have five (5) business days to cure such event.
(d) By Company for Other Than Cause. If the Company terminates Executive’s employment with the Company for any reason other than Cause, Executive shall be entitled to receive: (i) Accrued Compensation to the date of termination; (ii) severance compensation equal to Executive’s Base Compensation, immediately prior to the termination, for twelve (12) months after the date of termination (the “Termination Date”); (iii) continued participation in the Company medical and disability plans, at the Company’s expense, for twelve (12) months after the date of termination; and (iv) all insurance coverages, at the Company’s expense, in effect immediately prior to the termination. “Other Than Cause” shall include, but not be limited to the following: (i) without the Executive’s express written consent, the assignment to the Executive of any duties or the reduction of the Executive’s duties, either of which results in a significant diminution in the Executive’s position or responsibilities with the Company in effect immediately prior to such assignments or the removal of the Executive from such position and responsibilities; (ii) without the Executive’s express written consent, a substantial reduction, without good business reasons, of the facilities and perquisites (including office space and location) available to the Executive immediately prior to such reduction; (iii) a reduction by the Company in the Base Compensation of the Executive as in effect immediately prior to such reduction, other than a bonus reduction resulting from application of a bonus formula or plan on a basis that is consistent with prior practice; (iv) a material reduction by the Company in the kind or level of employee benefits to which the Executive is entitled immediately prior to such reduction with the result that the Executive’s overall benefits package is significantly reduced; (v) the relocation of the Executive to a facility or a location more than twenty-five (25) miles from the Executive’s then present location, without the Executive’s express written consent; (vi) any purported termination of the Executive by the Company which is not effected for Cause, for valid grounds, or due to the Executive’s death or Disability; or (vii) any purported termination of the Executive’s employment by the Company which is not effected pursuant to a notice of termination satisfying the requirements of Section I (b) above, and for purposes of this, Agreement, no such purported termination shall be effective. If Executive resigns due to one of the above enumerated factors, such resignation shall be deemed a termination by the Company for Other Than Cause and shall be governed by this Section 3(d).
(e) Insurance. If the Executive is entitled to severance compensation under Section 3(d), in addition to such severance benefits, the Executive shall receive health insurance coverage as provided to the Executive immediately prior to the Termination Date to the extent the cost of such health insurance coverage does not exceed the cost of such health insurance coverage prior to Executive’s termination. The health insurance coverage shall continue for a period of twelve (12) months following the Termination Date; provided, however, that to the extent Executive becomes covered under another employer’s group health insurance plan, the Company shall not be obligated to provide such health insurance coverage.
(f) No Duty to Mitigate. The Executive shall not be required to mitigate the amount of any payment contemplated by Section 3(d) (whether by seeking new employment or in any other manner).
4. Equity In the event Executive resigns due to one of the enumerated factors in Section 3(d), all of the unvested portion of the shares of Common Stock of the Company owned by the Executive pursuant to the Restricted Stock Purchase Agreement between the Company and the Executive dated June 22, 1998 shall immediately vest and be released from the Company’s repurchase option as of the Termination Date.
5. Confidential Information
(a) Company Information. Executive agrees at all times during the term of Executive’s employment and thereafter to hold in strictest confidence and not to use, except for the benefit of the Company, or to disclose to any person, firm or corporation without written authorization of the Board of Directors of the Company, any Confidential Information of the Company. Executive understands that “Confidential Information” means any Company proprietary information, technical data, trade secrets or know-how, including, but not limited to, research product plans, products, services, customer lists and customers (including, but not limited to, customers of the Company on whom Executive called or with whom Executive became acquainted during the term of Executive’s employment), markets, software, developments, inventions, processes, formulas, technology, designs, drawings, engineering, hardware configuration information, marketing, finances or other business information disclosed to Executive by the Company either directly or indirectly in writing, orally or by drawings or observation of parts or equipment. Executive further understands that Confidential Information does not include any of the foregoing item which has become publicly known and made generally available through no wrongful act of Executive’s or of others who were under confidentiality obligations as to the item or items involved.
(b) Former Employer Information. Executive agrees that Executive will not, during Executive’s employment with the Company, improperly use or disclose any proprietary information or trade secrets of any former or concurrent employer or other person or entity and that Executive will not bring onto the premises of the Company any unpublished document or proprietary information belonging to any such employer, person or entity unless consented to in writing by such employer, person or entity.
(c) Third Party Information. Executive recognizes that the Company has received and in the future will receive from third parties their confidential or proprietary information subject to a duty on the Company’s part to maintain the confidentiality of such information and to use it only for certain limited purposes. Executive agrees to hold all such confidential or proprietary information in the strictest confidence and not to disclose it to any person, firm or corporation or to use it except as necessary in carrying out his work for the Company consistent with the Company’s agreement with such third party.
6. Representations. Executive has not entered into, and will not enter into, any oral or written agreement in conflict herewith.
7. Arbitration. Executive and Company agree that any dispute or controversy arising out of or relating to any interpretation, construction, performance or breach of this Agreement shall be settled by arbitration of a single arbitrator to be held in South San Francisco, California, in accordance with the rules then in effect of the American Arbitration Association. The arbitrator may grant injunctions or other relief in such dispute or controversy. The decision of the arbitrator shall be final, conclusive and binding on the parties to the arbitration. Judgment may be entered on the arbitrator’s decision in any court having jurisdiction. The Company and Executive shall each pay one-half of the costs and expenses of such arbitration, and each party shall separately pay counsel fees and expenses.
8. General Provisions
(a) Governing Law; Consent to Personal Jurisdiction. This Agreement will be governed by the laws of the State of California. Executive and Company hereby expressly consent to the personal jurisdiction of the state and federal courts located in California for any lawsuit filed relating to this Agreement.
(b) Entire Agreement. This Agreement sets forth the entire agreement and understanding between the Company and Executive relating to the subject matter herein and merges all prior discussions between the parties. No modification of or amendment to this Agreement, nor any waiver of any rights under this Agreement, will be effective unless in writing signed by both parties hereto. Any subsequent change or changes in Executive’s duties, Salary or compensation will not affect the validity or scope of this Agreement.
(c) Severability. If one or more of the provisions in this Agreement are deemed void by law, then the remaining provisions will continue in fall force and effect.
(d) Successors and Assigns. This Agreement shall be binding upon and shall inure to the benefit of the parties and their respective heirs, legal representatives, successors, and permitted assigns.
(e) Counterparts. This Agreement may be executed in counterparts, each of which shall be deemed an original, but all of which together will constitute one and the some instruments.
IN WITNESS WHEREOF, each of the parties has executed this Agreement of the Company by its duly authorized OFFICER, as of the day and year first written above.
| REMI BARBIER, AN INDIVIDUAL | ||||
| Date: July 1 | /s/ REMI BARBIER | |||
| PAIN THERAPEUTICS, INC. | ||||
| Date: ______________________ | By: | /s/ REMI BARBIER | ||
| Title: | President & CEO | |||
[PAIN THERAPEUTICS, INC. LETTERHEAD]
AMENDMENT TO EMPLOYMENT AGREEMENT
This amendment (the “Amendment”) is made by and between Remi Barbier (the “Executive”) and Pain Therapeutics, Inc., a Delaware corporation (the “Company” and together with the Executive hereinafter collectively referred to as the “Parties”) on December 17, 2008.
WITNESSETH:
WHEREAS, the Parties previously entered into an employment agreement, date July 1, 1998 (the “Employment Agreement”); and
WHEREAS, the Company and Executive wish to amend the Employment Agreement, and bring certain terms into compliance with Section 409A of the Internal Revenue Code and the final regulations and other official guidance thereunder (“Section 409A”), as set forth below.
NOW, THEREFORE, for good and valuable consideration, Executive and the Company agree that the Employment Agreement is hereby amended as follows:
1. The following language replaces the first clause (iii) in Section 3(d) of the Employment Agreement in its entirety:
“(iii) continued participation in the Company medical plan, at the Company’s expense, for twelve (12) months after the date of termination”
2. The following language replaces the first clause (iv) in Section 3(d) of the Employment Agreement in its entirety:
“(iv) a lump-sum payment which is, on an after-tax basis, for the Executive sufficient to provide Executive with no less than equivalent coverage under third-party plans for a period of twelve (12) months for all insurance coverage’s (including disability insurance coverage) in effect immediately prior to termination.”
3. The following language is inserted immediately following Section 3(f) of the Employment Agreement and shall constitute Section 3(g) of the Employment Agreement:
“(g) 409A Compliance. Notwithstanding anything to the contrary in this Agreement, no Deferred Payments (as defined below) will be considered due or payable until the Executive has incurred a “separations from service” within the meaning of Section 409A.
Notwithstanding anything to the contrary in this Agreement, no severance pay or benefits payable upon separation, that are payable to the Executive, if any, pursuant to this Agreement, when considered together with any other severance payments or separation benefits that are considered deferred compensation (together, the “Deferred Payments”) under Section 409A will be payable until the Executive has a “separation from service” within the meaning of Section 409A.
Notwithstanding anything to the contrary in this Agreement, if the Executive is a “specified employee” within the meaning of Section 409A at the time of the Executive’s termination of employment, then, if required, the Deferred Payments, which are otherwise due to the Executive on or within the 6 month period following the Executive’s termination will accrue, to the extent required, during such 6 month period and will become payable in a lump sum payment on the date 6 months and 1 day following the date of the Executive’s termination of employment or the date of the Executive’s death, if earlier. All subsequent Deferred Payments, if any, will be payable in accordance with the payment schedule applicable to each payment or benefit. Each payment and benefit payable under this Agreement is intended to constitute a separate payment for purposes of Section 1.409A-2(b)(2) of the Treasury Regulations.
The foregoing provision is intended to comply with the requirements of Section 409A so that none of the severance payments and benefits to be provided hereunder will be subject to the additional tax imposed under Section 409A, and any ambiguities herein will be interpreted to so comply. The Executive and the Company agree to work together in good faith to consider amendments to this Agreement and to take such reasonable actions which are necessary, appropriate or desirable to avoid imposition of any additional tax or income recognition prior to actual payment to the Executive under Section 409A.”
These provisions are not intended to increase or to decrease any severance or other compensation arrangements currently in place between you and the Company.
(Signature page follows)
IN WITNESS WHEREOF, each of the Parties has executed this Amendment, in the case of the Company by its duly authorized officer, as of the day and year set forth above.
| Pain Therapeutics, Inc. | Executive | |||||||
| By: | By: | |||||||
| Name: | Peter S. Roddy | Remi Barbier | ||||||
| Title: | Vice President & CFO | |||||||
EXHIBIT 10.13
[PAIN THERAPEUTICS, INC. LETTERHEAD]
August 29, 2000
Grant L. Schoenhard, Ph.D.
[address]
Dear Grant:
Barry Sherman and I are very pleased to offer you the position of Vice President, Preclinical Development at Pain Therapeutics, Inc. We believe this offer reflects both the letter and the spirit of previous discussions. Terms of your offer of employment are outlined below:
1. As Vice President, Preclinical Development, you will initially report to Barry Sherman, MD, PTI’s Executive Vice President & Chief Medical Officer.
2. Your primary responsibilities will include providing PTI with preclinical and clinical pharmacology support for PTI’s entire pipeline of products. This role is crucial to PTI’s regulatory and clinical groups to assure the timely, successful completion of PTI’s clinical programs.
3. Your cash compensation will be $175,000 per year and will be reviewed annually. In addition, PTI will reimburse you for all reasonable business and travel expenses actually incurred on behalf of PTI.
4. You will receive an option to buy 50,000 shares of PTI common stock. All options are priced at the fair market value of PTI’s common stock at the date of grant. Your option will vest monthly and equally over 48 months, starting on the start date of your full-time employment by PTI.
5. Your mutually agreed upon start date is Monday, September 11, 2000.
6. You will be eligible to receive medical, life insurance, disability or other health, insurance and other benefits provided to regular full-time PTI.
7. You will be entitled to three (3) weeks paid vacation at times mutually agreeable to you and PTI Vacation time is accrued at the rate of 1.25 days per month. Unused vacation may not be reimbursed or carried forward from year to year.
8. You acknowledge and agree that in accordance with California law, your employment at PTI is “at will”. You understand that PTI or you may terminate your employment at any time, for any reason or no reason, with or without cause and with and without notice. PTI also reserves the right to make personnel decisions regarding your employment, including but not limited to, promotions, salary adjustment, scope of responsibilities, transfer and termination consistent with PTI’s needs.
In the event PTI terminates your employment without cause after your one year anniversary, PTI will continue to provide you with your regular base salary and health benefits until the earlier of a) three months from date of termination, or b) your date of new employment or other compensated position elsewhere. You will not receive severance or other termination benefits or any other benefits (including vesting of unvested stock) in the event either a) you terminate this employment arrangement for any reason or no reason, or b) PTI terminates this employment arrangement for any reason or no reason in the first 12 months of your full-time employment, or c) PTI terminates this employment arrangement with cause.
9. You and PTI further agree that all disputes, claims or causes of action arising out of your employment or its termination shall be submitted to binding arbitration before a neutral arbitrator, except where the law specifically forbids the use of arbitration as a final and binding remedy.
10. You warrant and represent that you have no commitments or obligations inconsistent with PTI’s offer of employment as of the date of your full-time employment with PTI. You further understand that this is a full-time and exclusive position in the services of PTI.
11. You agree to sign a CONFIDENTIAL INFORMATION AND INVENTION ASSIGNMENT AGREEMENT (attached).
12. This offer expires Thursday, August 31, 2000 unless signed by you and received by PTI before then.
Grant, I believe these terms reflect our discussions. If acceptable to you, please sign, date and return one original copy. We look forward to working with you!
| /s/ REMI BARBIER |
| Remi Barbier President & CEO |
I agree to all the terms and condition of employment set forth in this letter,
| /s/ GRANT L. SCHOENHARD |
| Grant L. Schoenhard |
AMENDMENT TO OFFER LETTER
This amendment (the “Amendment”) is made by and between Grant L. Schoenhard (the “Executive”) and Pain Therapeutics, Inc., a Delaware corporation (the “Company” and together with the Executive hereinafter collectively referred to as the “Parties”) on December 30, 2008.
WITNESSETH:
WHEREAS, the Parties previously entered into an offer letter, date August 29, 2000 (the “Offer Letter”); and
WHEREAS, the Company and Executive wish to amend the Offer Letter, and bring certain terms into compliance with Section 409A of the Internal Revenue Code and the final regulations and other official guidance thereunder (“Section 409A”), as set forth below.
NOW, THEREFORE, for good and valuable consideration, Executive and the Company agree that the Offer Letter is hereby amended as follows:
| 1. | The following language is inserted immediately following the second full paragraph on the second page of the Offer Letter: |
“Notwithstanding anything to the contrary in this offer letter, no Deferred Payments (as defined below) will be considered due or payable until you have incurred a “separation from service” within the meaning of Section 409A of the Internal Revenue Code of 1986, as amended and the final regulations and any guidance promulgated thereunder (together, “Section 409A”).
Notwithstanding anything to the contrary in this offer letter, no severance pay or benefits payable upon separation, that are payable to you, if any, pursuant to this offer letter, when considered together with any other severance payments or separation benefits that are considered deferred compensation (together, the “Deferred Payments”) under Section 409A will be payable until you have a “separation from service” within the meaning of Section 409A.
Notwithstanding anything to the contrary in this offer letter, if you are a “specified employee” within the meaning of Section 409A at the time of your termination of employment, then, if required, the Deferred Payments, which are otherwise due to you on or within the 6 month period following your termination will accrue, to the extent required, during such 6 month period and will become payable in a lump sum payment on the date 6 months and 1 day following the date of your termination of employment or the date of your death, if earlier. All subsequent Deferred Payments, if any, will be payable in accordance with the payment schedule applicable to each payment or benefit. Each payment and benefit payable under this offer letter is intended to constitute a separate payment for purposes of Section 1.409A-2(b)(2) of the Treasury Regulations.
The foregoing provision is intended to comply with the requirements of Section 409A so that none of the severance payments and benefits to be provided hereunder will be subject to the additional tax imposed under Section 409A, and any ambiguities herein will be interpreted to so comply. You and the Company agree to work together in good faith to consider amendments to this offer letter and to take such reasonable actions which are necessary, appropriate or desirable to avoid imposition of any additional tax or income recognition prior to actual payment to you under Section 409A.”
These provisions are not intended to increase or to decrease any severance or other compensation arrangements currently in place between you and the Company.
(Signature page follows)
IN WITNESS WHEREOF, each of the Parties has executed this Amendment, in the case of the Company by its duly authorized officer, as of the day and year set forth above.
| Pain Therapeutics, Inc. | Executive | |||||||
| By: | By: | |||||||
| Name: | Remi Barbier | Grant L. Schoenhard | ||||||
| Title: | President & CEO | |||||||
EXHIBIT 10.14
[PAIN THERAPEUTICS, INC. LETTERHEAD]
November 5, 2002
Mr. Peter S. Roddy
[address]
Dear Pete:
I believe PTI could benefit tremendously from your years of professional experience working in the life science industry. In this spirit, I am pleased to offer you the position of Chief Financial Officer at Pain Therapeutics, Inc. This is an officer level position reporting directly to Remi Barbier, Chairman of the Board, President and Chief Executive Officer.
Terms of your offer of employment are outlined below:
| 1. | Your primary responsibilities are listed below, with an understanding that other responsibilities will arise from time to time: |
| • | Prepare and maintain PTI’s internal financial operations, including its finance, accounting, budgeting and administrative functions; |
| • | Prepare, file and maintain PTI’s external financial reporting requirements, including making all personal certifications, as required by the Securities and Exchange Commission, and other regulatory bodies, and by Sarbanes-Oxley Act of 2002 and related requirements; |
| • | Provide financial strategy, guidance and support to PTI’s long-term strategic planning efforts. |
| 2. | Your cash compensation will be $220,000 (two hundred twenty thousand) per year, and shall be payable semi-monthly in accordance with PTI’s standard payroll practices. In addition, PTI will reimburse you for all reasonable business and travel expenses actually incurred on behalf of PTI. |
Your performance and your compensation will be reviewed at such time as PTI generally conducts an annual review of all company officers (which generally occurs in July of each year). You will be eligible to receive a discretionary cash and/or equity bonus at the time of your performance review.
| 3. | In addition, if you decide to join PTI, it will be recommended at the first meeting of the company’s Board of Directors following your start date that PTI grant you an option to purchase 200,000 (two hundred thousand) shares of the company’s common stock at a price per share equal to the closing price for one share of the company’s common stock, as reported by NASDAQ, on the last market trading day prior to the date of the grant of such option. Your option will vest monthly and equally over 48 months. Your option will begin vesting on the first day of your full-time employment by PTI. This option shall be subject to the terms and conditions of the 1998 Stock Plan and standard form of stock option agreement, including vesting and exercise requirements. No right to any stock is earned or accrued until such time that vesting occurs, nor does the grant confer any right to continue vesting or employment. |
| 4. | You hereby agree that your full-time start date will be Monday, November 18, 2002. |
| 5. | You will be eligible to receive medical, life insurance, disability or other health, insurance and other benefits provided to regular full-time PTI. The details of the benefits are outlined in Exhibit A hereto. |
| 6. | You will be entitled to three (3) weeks of paid vacation per year. PTI Vacation time is accrued at the rate of 1.25 days per month of full-time employment. Accrued but unused vacation beyond 15 days may not be reimbursed or carried forward from year to year. |
| 7. | You acknowledge and agree that in accordance with California law, your employment at PTI is “at will”. You understand that PTI or you may terminate your employment at any time, for any reason or no reason, with or without cause and with and without notice. PTI also reserves the right to make personnel decisions regarding your employment, including but not limited to, promotions, salary adjustment, scope of responsibilities, transfer and termination consistent with PTI’s needs. |
In the event PTI terminates your employment without cause after your one year anniversary, PTI will continue to provide you with your regular base salary and health benefits until the earlier of a) three months from date of termination, or b) your date of new employment or other compensated position elsewhere. You will not receive severance or other termination benefits or any other benefits (including vesting of unvested stock) in the event either a) you terminate this employment arrangement for any reason or no reason, or b) PTI terminates this employment arrangement for any reason or no reason in the first 12 months of your full-time employment, or c) PTI terminates this employment arrangement with cause.
| 8. | For purposes of federal immigration law, you will be required to provide to the Company documentary evidence of your identify and eligibility for employment in the United States. Such documentation must be provided to us within three (3) business days of your date of hire, or our employment relationship with you may be terminated. |
| 9. | We also ask that, if you have not already done so, you disclose to us any and all agreements relating to your prior employment that may affect your eligibility to be employed by the Company or limit the manner in which you may be employed. It is PTI’s understanding that any such agreements will not prevent you from performing the duties of your position and you represent that such is the case. Moreover, you agree that, during the term of your employment with PTI, you will not engage in any other employment, occupation, consulting or other business activity directly related to the business in which PTI is now involved or becomes involved during the term of your employment, nor will you engage in any other activities that conflict with your obligations to PTI. Similarly, you agree not to bring any third party confidential information to PTI, including that of your former employer, and that in performing your duties for PTI you will not in any way utilize any such information. |
| 10. | As an employee of PTI, you will be expected to abide by the Company’s rules and standards. Specifically, you will be required to sign an acknowledgment that you have read and that you understand the Company’s rules of conduct which are included in the Company Handbook and that you have no commitments or obligations inconsistent with PTI’s offer of employment as of the date of your full-time employment with PTI. You further understand that this is a full-time and exclusive position in the services of PTI. |
| 11. | As a condition to your employment, you agree to sign the attached CONFIDENTIAL INFORMATION AND INVENTION ASSIGNMENT AGREEMENT, which requires, among other provisions, the assignment of patent rights to any invention made during your employment at the Company, and non-disclosure of Company proprietary information. |
| 12. | In the event of any dispute or claim relating to or arising out of our employment relationship, you and PTI hereby agree that (i) any and all disputes between you and the Company shall be fully and finally resolved by binding arbitration, (ii) you are waiving any and all rights to a jury trial but all court remedies will be available in arbitration, (iii) all disputes shall be resolved by a neutral arbitrator who shall issue a written opinion, (iv) the arbitration shall provide for adequate discovery, and (v) the Company shall pay all but the first $200 of the arbitration fees. Please note that we must receive your signed Agreement before your first day of employment. |
To accept this offer, please sign and date this letter in the space provided below. A duplicate original is enclosed for your records. If you accept our offer, your first day of employment will be November 18, 2002. This letter, along with any agreements relating to proprietary rights between you and PTI, set forth the terms of your employment with the Company and supersede any prior representations or agreements including, but not limited to, any representations made during your recruitment, interviews or pre-employment negotiations, whether written or oral. This letter, including, but not limited to, its at-will employment provision, may not be modified or amended except by a written agreement signed by you and I. This offer of employment will terminate if it is not accepted, signed and returned by November 17, 2002.
Pete, we look forward to your favorable reply and to working with you at PTI.
| Sincerely, |
| /s/ REMI BARBIER |
| Remi Barbier Chairman of the Board, President & CEO |
| Agreed to and accepted: | ||
| Signature: | /s/ PETER S. RODDY | |
| Printed Name: | Peter S. Roddy | |
| Date: | 11/18/2008 | |
Enclosures
Duplicate Original Letter
Confidential Information and Invention Assignment Agreement
AMENDMENT TO OFFER LETTER
This amendment (the “Amendment”) is made by and between Peter S. Roddy (the “Executive”) and Pain Therapeutics, Inc., a Delaware corporation (the “Company” and together with the Executive hereinafter collectively referred to as the “Parties”) on December 30, 2008.
WITNESSETH:
WHEREAS, the Parties previously entered into an offer letter, dated November 5, 2002 (the “Offer Letter”); and
WHEREAS, the Company and Executive wish to amend the Offer Letter, and bring certain terms into compliance with Section 409A of the Internal Revenue Code and the final regulations and other official guidance thereunder (“Section 409A”), as set forth below.
NOW, THEREFORE, for good and valuable consideration, Executive and the Company agree that the Offer Letter is hereby amended as follows:
| 1. | The following language is inserted immediately following the second full paragraph on the second page of the Offer Letter: |
“Notwithstanding the foregoing, bonus payments payable hereunder shall in no event be paid after the later of (i) 2 1/2 months after the end of PTI’s fiscal year in which such bonus is earned, or (ii) March 15 following the calendar year in which such bonus is earned.”
| 2. | The following language is inserted immediately following the first full paragraph on the third page of the Offer Letter: |
“Notwithstanding anything to the contrary in this offer letter, no Deferred Payments (as defined below) will be considered due or payable until you have incurred a “separation from service” within the meaning of Section 409A.
Notwithstanding anything to the contrary in this offer letter, no severance pay or benefits payable upon separation, that are payable to you, if any, pursuant to this offer letter, when considered together with any other severance payments or separation benefits that are considered deferred compensation (together, the “Deferred Payments”) under Section 409A will be payable until you have a “separation from service” within the meaning of Section 409A.
Notwithstanding anything to the contrary in this offer letter, if you are a “specified employee” within the meaning of Section 409A at the time of your termination of employment, then, if required, the Deferred Payments, which are otherwise due to you on or
within the 6 month period following your termination will accrue, to the extent required, during such 6 month period and will become payable in a lump sum payment on the date 6 months and 1 day following the date of your termination of employment or the date of your death, if earlier. All subsequent Deferred Payments, if any, will be payable in accordance with the payment schedule applicable to each payment or benefit. Each payment and benefit payable under this offer letter is intended to constitute a separate payment for purposes of Section 1.409A-2(b)(2) of the Treasury Regulations.
The foregoing provision is intended to comply with the requirements of Section 409A so that none of the severance payments and benefits to be provided hereunder will be subject to the additional tax imposed under Section 409A, and any ambiguities herein will be interpreted to so comply. You and the Company agree to work together in good faith to consider amendments to this offer letter and to take such reasonable actions which are necessary, appropriate or desirable to avoid imposition of any additional tax or income recognition prior to actual payment to you under Section 409A.”
These provisions are not intended to increase or to decrease any severance or other compensation arrangements currently in place between you and the Company.
(Signature page follows)
IN WITNESS WHEREOF, each of the Parties has executed this Amendment, in the case of the Company by its duly authorized officer, as of the day and year set forth above.
| Pain Therapeutics, Inc. | Executive | |||||||
| By: | By: | |||||||
| Name: | Remi Barbier | Peter S. Roddy | ||||||
| Title: | President & CEO | |||||||
EXHIBIT 23.1
CONSENT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
We consent to the incorporation by reference in the Registration Statement (Form S-3 No. 333-115362) of Pain Therapeutics, Inc. and in the related Prospectus, and in the Registration Statements (Form S-8 Nos. 333-147336, 333-134364, 333-115361, 333-105138, 333-68118 and 333-152676) pertaining to the 2008 Equity Incentive Plan, the 1998 Stock Plan and 2000 Employee Stock Purchase Plan of Pain Therapeutics, Inc. of our reports dated February 11, 2009, with respect to the financial statements of Pain Therapeutics, Inc., and the effectiveness of internal control over financial reporting of Pain Therapeutics, Inc., included in this Annual Report (Form 10-K) for the year ended December 31, 2008.
/s/ ERNST & YOUNG LLP
San Jose, California
February 11, 2009
EXHIBIT 31.1
PRINCIPAL EXECUTIVE OFFICER CERTIFICATION PURSUANT TO
SECTION 302 OF THE SARBANES-OXLEY ACT OF 2002
I, Remi Barbier, certify that:
| 1. | I have reviewed this Report on Form 10-K of Pain Therapeutics, Inc.; |
| 2. | Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report; |
| 3. | Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report; |
| 4. | The registrant’s other certifying officer(s) and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have: |
| a. | Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared; |
| b. | Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles; |
| c. | Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and |
| d. | Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and |
| 5. | The registrant’s other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the registrant’s board of directors (or persons performing the equivalent functions): |
| a. | All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and |
| b. | Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting. |
| /s/ REMI BARBIER |
| Remi Barbier, |
| Chairman of the Board of Directors, President and Chief Executive Officer (Principal Executive Officer) |
Date: February 11, 2009
EXHIBIT 31.2
PRINCIPAL FINANCIAL OFFICER CERTIFICATION PURSUANT TO
SECTION 302 OF THE SARBANES-OXLEY ACT OF 2002
I, Peter S. Roddy, certify that:
| 1. | I have reviewed this Report on Form 10-K of Pain Therapeutics, Inc.; |
| 2. | Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report; |
| 3. | Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report; |
| 4. | The registrant’s other certifying officer(s) and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have: |
| a. | Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared; |
| b. | Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles; |
| c. | Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and |
| d. | Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and |
| 5. | The registrant’s other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the registrant’s board of directors (or persons performing the equivalent functions): |
| a. | All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and |
| b. | Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting. |
| /s/ PETER S. RODDY |
| Peter S. Roddy, |
| Vice President and Chief Financial Officer (Principal Financial Officer) |
Date: February 11, 2009
EXHIBIT 32.1
CERTIFICATIONS OF THE CHIEF EXECUTIVE OFFICER AND THE
CHIEF FINANCIAL OFFICER PURSUANT TO 18 U.S.C. SECTION 1350,
AS ADOPTED PURSUANT TO SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
Pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, Remi Barbier, Chairman of the Board of Directors, President and Chief Executive Officer and Peter S. Roddy, Vice President and Chief Financial Officer of Pain Therapeutics, Inc. (the “Company”), hereby certify that to the best of our knowledge:
| 1. | The Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2008, and to which this certification is attached as Exhibit 32.1 (the “Periodic Report”), fully complies with the requirements of Section 13 (a) or 15 (d) of the Securities Exchange Act of 1934, and |
| 2. | The information contained in this Periodic Report fairly presents, in all material respects, the financial condition and results of operations of the Company. |
| Date: February 11, 2009 |
| /S/ REMI BARBIER |
| Remi Barbier, |
| Chairman of the Board of Directors, President and Chief Executive Officer |
| /S/ PETER S. RODDY |
| Peter S. Roddy, Vice President and Chief Financial Officer |